


1. Introduction
Bacteriophages are the most abundant biological entity on Earth with an estimated 1031 particles being present on our planet. Among these, tailed bacteriophages with double-stranded DNA genomes are the most dominantly observed and reported in the literature [1]. Until recently, tailed phages were classified within the Caudovirales order and described based on their morphology, possessing either a short (Podoviridae) or long tail that is contractile (Myoviridae) or non-contractile (Siphoviridae). Given the extent of their genetics, host species, and ecological diversity, the taxonomy of phages has recently been re-established to reflect these evolutionarily relevant factors and to reduce reliance on morphology-based assignments [2]. The current viral class Caudoviricetes encompasses 14 families assigned to one of four orders, while a further 33 families have been identified that have yet to be assigned to an order. Irrespective of the (sequence-based) taxonomy of phages, their morphology has a significant bearing on the types of interactions they will establish with their respective hosts [3,4].
Phages with long, non-contractile tails (formerly called the Siphoviridae) are the most abundant in nature and several of these have become models to study phage–host interactions, including the Lactococcus phages TP901-1 and p2, Streptococcus thermophilus phage STP1, Bacillus subtilis phage SPP1, and Escherichia coli phages lambda and T5 [5,6,7,8,9,10]. These phages are genetically distinct; however, in many cases, functional modules within their genomes are syntenic [11]. Furthermore, despite sequence disparity between phages of the same or distinct bacterial host species, the structure of their components is often conserved [4,12,13,14,15]. The temperate lactococcal P335 phage TP901-1 is particularly well characterized with respect to its three-dimensional structure and the interactions with its host [5,16,17,18]. Furthermore, it has been employed as a model to understand the phenomenon of lysogeny and the factors that underpin the lytic–lysogenic lifestyle decision process [19,20,21]. TP901-1 structural module encompassing 22 genes that are responsible for the biosynthesis of the phage head, tail, baseplate, and DNA packaging machinery (Figure 1).
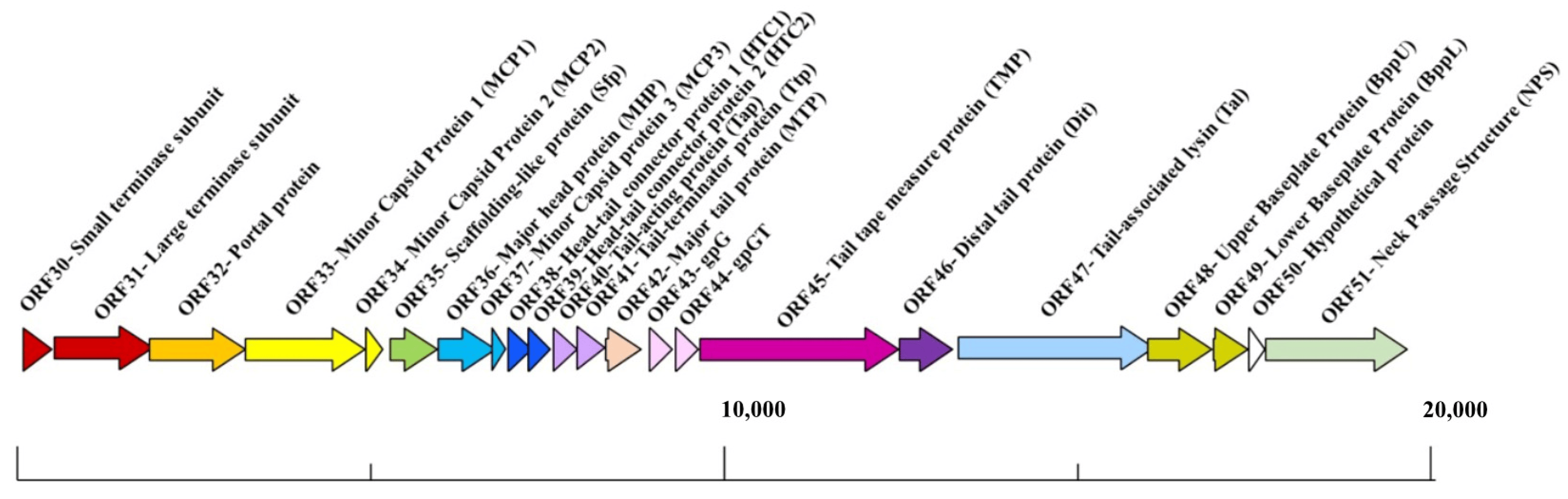
Figure 1. Schematic representation of the structural module of TP901-1. The functions are indicated above the arrows and the scale bar is presented at the base of the schematic, measured in base pairs (bp). This figure and associated functions are based on those described in reference [17].
TP901-1 possesses an isometric capsid and a long non-contractile tail of approximately 135 nm [22]. At the distal end of its tail, TP901-1 presents a large, multi-protein adhesion device, termed a baseplate, which is composed of the tail tape measure protein (TMP), distal tail protein (Dit), tail-associated lysin (Tal), and upper (BppU) and lower (BppL) baseplate proteins [5,23]. The BppL component is the bona fide receptor-binding protein (RBP) and presents as 18 trimers in the whole baseplate structure, which likely recognize and bind to cell wall polysaccharides present on the surface of the host lactococcal cell. Its exquisite host specificity is based on its heterologous expression of the Lactococcus lactis genetic region, encoding host-specific glycosyltransferases associated with cell wall polysaccharide biosynthesis [24].
The adhesion device of TP901-1 is in an infection-ready conformation and does not require activation by divalent cations, such as calcium, as is the case for certain tailed phages including the lactococcal phage p2 [25]. Mutational analyses of the TP901-1 gene coding for the capsid and tail proteins have provided insights into the assembly of the mature virion and the function of several previously uncharacterized gene products [17]. Furthermore, detailed genetic and structural analyses of the tail and baseplate proteins have rendered TP901-1 one of the best-characterized phages capable of infecting Gram-positive bacteria [5,22,26].
The recent development of AlphaFold2 (AF2) has significantly enhanced and advanced our capabilities to predict individual and multi-protein structures [27,28,29] and has transformed the field of structural biology. The ever-increasing number of phage genome and protein sequences and protein structures in public databases reinforces the need of reliable and rapid methods for the determination of (multi-component) protein structures and the associated protein functionality. In the present study, we present predicted structures of the TP901-1 capsid and neck, its extended tail, and its complete baseplate structure, which was previously partially determined with X-ray crystallography. AF2 reliably predicted the structures of large parts of the phage in most cases yet was unsuccessful in a small number of cases, in which chaperone-aided folding was probably the reason for such structure prediction failure.
2. Materials and Methods
We performed predictions using either a Colab notebook running AlphaFold v2.3.1 (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb, 1 January 2023) on Nvidia A100 40 Gb GPU, or HPC resources from GENCI-IDRIS running AlphaFold v2.3.1 on a Jean Zay supercomputer A100 80Gb GPU. The retry number was set to 20 and no relaxation was performed. Five models were generated for each prediction. Colab was used with small complexes while Jean Zay was used for large complexes requiring longer calculation times.
The virion structure of the tailed phage that we present here is one of the most complete to date. For such prediction-based structural work, the validity of the predicted structures is an important issue. AF2 has been recognized to provide high-quality structures provided that the pLDDT values are sufficiently high, i.e., above 80% [27,28,29,30]. Here, we checked the structure’s validity at three levels. First, internal to AF2, we only accepted structures that met the AF2 internal validation score given by the pLDDT values, i.e., when the pLDDT values were above 70% in the folded regions. Although loops often occur below this threshold, they do not significantly affect the overall fold. Moreover, we also examined the PAE plots, which give confidence scores of protein–protein interactions, and verified that the pLDDT values in the protein complex were at least equal or higher than those of the non-complexed structure. Second, we used an external validity criterion by assessing the agreement between the predicted structures and the experimental TP901-1 nsEM 3D reconstructions in the 15–25 Å-resolution range. We also used the available crystal structures to evaluate the capabilities and limitations of AF2’s predictions. As a third validation approach, we checked with the Dali server [31] whether the predicted folds had already been reported in the PDB. The plots of the pLDDT values, stored in the PDB file as B-factors, as well as the PAEs are shown in . Additionally, we used the PISA server [32] to analyze the quality of the interactions within our assemblies.
The pLDDT values of the predicted structures, stored in the PDB file as B-factors, as well as the PAEs were plotted and are shown in the . The final predicted protein or domain structures were submitted to the Dali server [31] to identify their closest structural homologs in the PDB. Visual representations of the structures were prepared with ChimeraX [33]. Coot [34,35] was used to assemble and visually analyze the predictions.
3. Results
3.1. The TP901-1 Capsid
Phage capsids are robust containers that carry and protect the viral genome packaged within its internal cavity [36]. Previously, the phage TP901-1 capsid structure has been determined via negative staining electron microscopy (nsEM) and single-particle analyses applying icosahedral symmetry at a 15 Å resolution [22]. The mature capsid (660 Å wide) assembles 60 hexamers (hexons) and 11 pentamers (pentons) of the ORF36 major capsid protein (MCP), with a T = 7 symmetry (EMD-2133). The 12th penton is replaced by a dodecamer of the portal protein. Here, we predicted the structures of these MCP hexamers and pentamers with confident scores, except for the N-terminus residues 1 to 15 (Figure 2, ). In both assemblies, the capsid MCP monomer contains the classical domains of a phage capsid protein, including, from the N-terminus to the C-terminus, the β-hairpin “E-loop”, the “P-domain” β-sheet, and a central “A-domain” formed of β-strands 4–6 and α-helices 3–5 (Figure 2A,C). While the A-domains form compact structures in the hexons and pentons, the looser E-loops cover an adjacent monomer (Figure 2B,D).
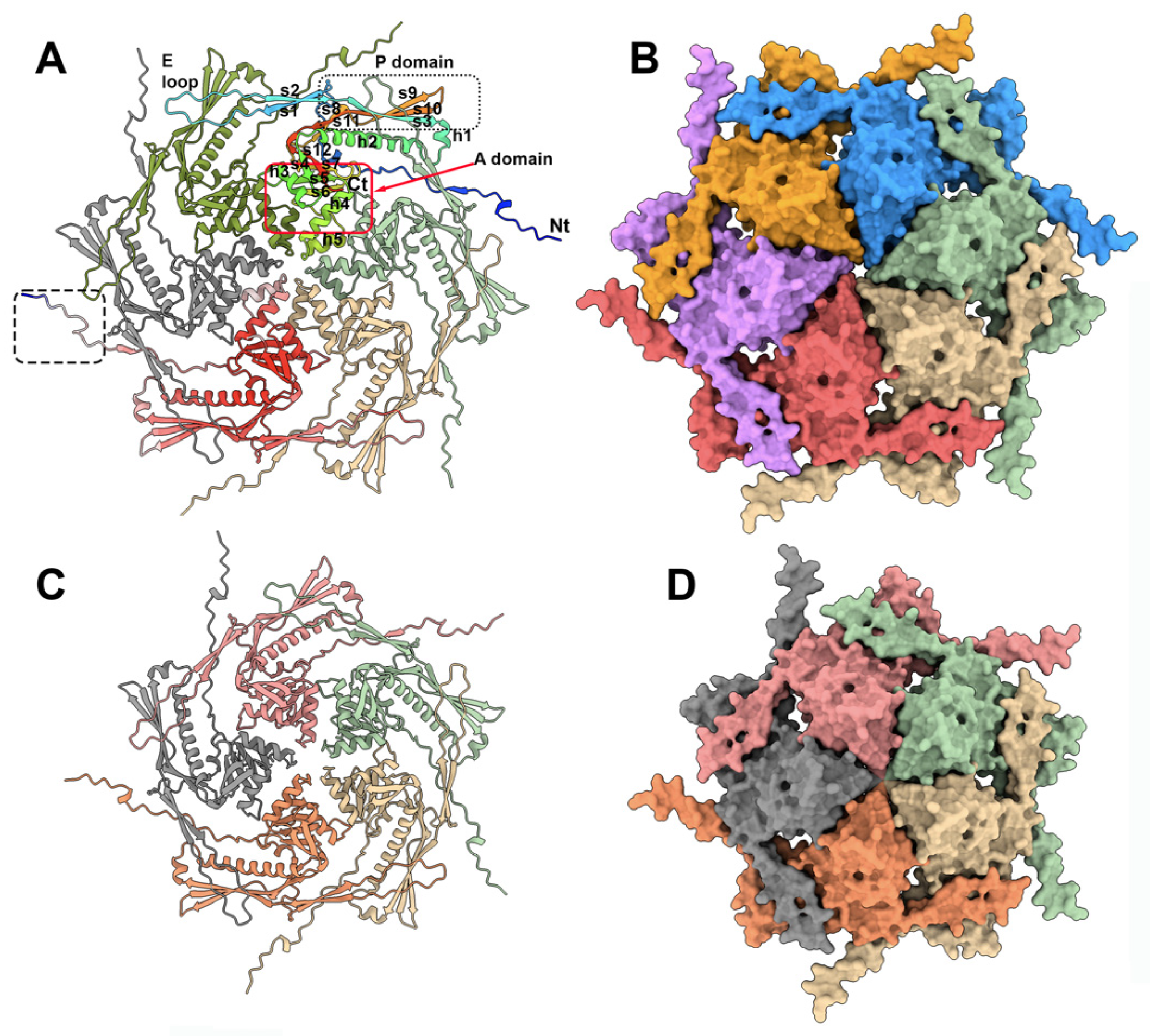
Figure 2. TP901-1 capsid’s hexon and penton structures. (A) Ribbon view of the predicted hexon structure. One monomer is rainbow colored and its domains are labeled. The monomer at the bottom of the figure is colored according to the pLDDT values (the quality of the prediction), from blue (low) to red (high). Note the low-confidence structure of the N-terminus (bottom dashed box). (B) Surface representation of the hexon (same orientation and scale as in (A)). (C) Ribbon view of the predicted penton structure. (D) Surface representation of the penton (same orientation and scale as in (C)).
In the entire capsid, each penton is surrounded by five hexons. Therefore, in order to analyze the protein contacts at the hexon–hexon and hexon–penton interfaces, we attempted to predict the structures of two hexons and of one hexon with one penton by feeding AF2 with 12 and 11 MCP sequences, respectively. However, in both cases, AF2 returned the hexons and penton separated, thus limiting the possibility to examine the interfaces. In order to overcome this limitation and to evaluate the predictions in the context of the whole capsid, we fitted the two predicted hexons and one penton (in the absence of residues 1–15) in the capsid nsEM 3D reconstruction (cross-correlation values of 0.81 (hexon) and 0.78 (penton)) . The TP901-1 capsid is structurally very similar to the coliphage HK97 [37], as reported by Dali (PDB ID: 1ohg; Z-score = 18.1; rmsd = 3.0 Å, for 248 aligned residues of the 280 residues in total).
3.2. The Procapsid Scaffolding Protein
Procapsids (virion precursors without DNA) are assembled from the dodecameric portal via the addition of MCP hexons and pentons [38]. This process is promoted by scaffolding proteins that have been proposed to establish bridges between capsid MCPs [39,40]. Little is known, however, about the structure of these scaffolding proteins, either alone or when they are attached to the MCPs. The procapsid structure of the Staphylococcus aureus phage 80α reveals the presence of an α-helix attached to each MCP on the internal face of the procapsid [39] (Figure 3A,B). This short α-helix, which belongs to the C-terminal end of the scaffolding protein, is only a small portion of the phage 80α’s full-length scaffolding protein (209 residues). The putative scaffolding protein of TP901-1 is slightly longer than that of 80α at its C-terminus (220 residues) (Figure 3B). Therefore, we predicted their structures, which assemble dimers, with good statistics (Figure 3C). However, while the 80α’s scaffolding protein shows an unstructured C-terminal end, that of TP901-1 shows three well-folded C-terminal α-helices (Figure 3D). We also predicted the structure of the complex between these C-terminal helices and an MCP hexon, and we found that each MCP is bound to this helical domain on the internal face of the hexon (Figure 3E,F). Scaffolding proteins (SPs) bind to the MCPs from the non-mature capsid and are released upon maturation [39]. Here, the MCP to which the SP binds resembles MCPs from mature capsids, indicating that AF2 is unable to discriminate between non-mature and mature MCPs for SP binding.
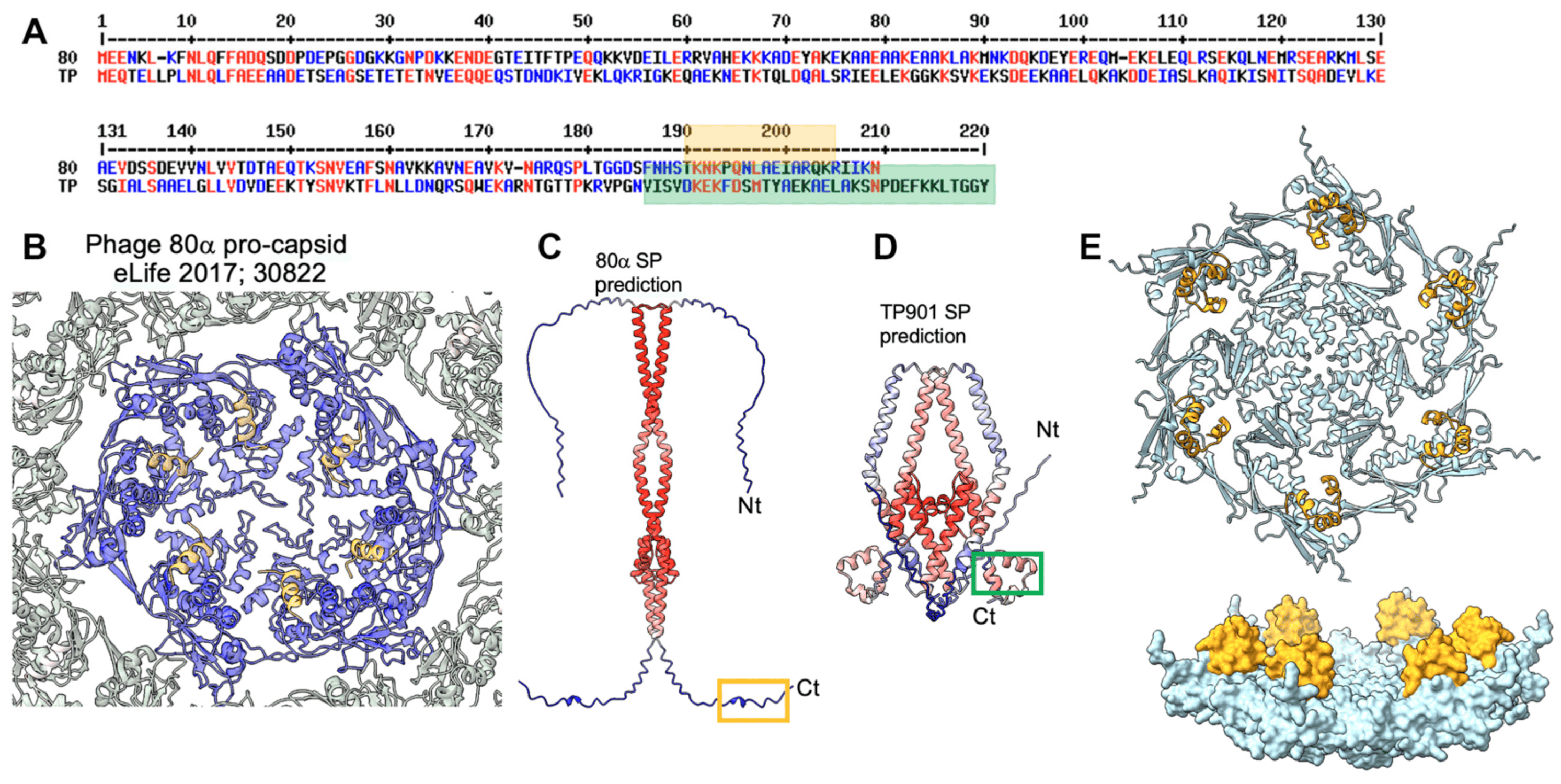
Figure 3. The procapsid’s scaffolding proteins (SPs). (A) Amino acid alignment between the scaffolding proteins of staphylococcal phage 80α and lactococcal phage TP901-1. (B) Ribbon representation of the cryoEM structure of a phage 80α MCP hexamer (blue) in complex with the C-terminus of the scaffolding protein (orange; see also the orange box in (A)) [1]. (C) AF2 prediction of the full-length phage 80α scaffolding protein. The orange box corresponds to the orange helix in (B). (D) AF2 prediction of the full-length phage TP901-1 scaffolding protein. The green-boxed helices correspond to the green box in (A). (E, top) Ribbon representation of a phage TP901-1 MCP hexamer (light blue) in complex with the C-terminus of the scaffolding protein (orange; see also the green box in (A)). (E, bottom) Surface view of the same structure rotated 90°.
3.3. Dodecameric Portal and Adaptor and the Hexameric Stopper
3.4. The Neck/Tail Junction and the Tail
The structures of several protein complexes were predicted for the neck/tail junction and the tail: (i) the complex formed by the adaptor (minus its C-terminal helix), stopper, tail terminator (TT), and MTP (12/6/6/6 mer); (ii) the adaptor/stopper complex (12/6 mer); (iii) the stopper/TT complex (6/6 mer); (iv) the TT/MTP complex (6/6 mer); and (v) the MTP/MTP complex (6/6 mer).
3.5. The Baseplate
The baseplate of TP901-1 comprises four proteins: the Dit, Tal, BppU, and RBP (ORF46, 47, 48, and 49, respectively) [5,11,50,51]. The Dit hexamer and the Tal trimer form the central part of the baseplate and extend the last MTP ring out of the tail. Moreover, the tape measure protein (TMP; ORF45) has been proposed to fill the central channel of the tail, Dit, and Tal [26]. A crystal structure of the TP901-1 baseplate, without the Tal, has previously been reported [5]. In this complex, six BppU trimers are attached to the Dit hexamer, each trimer projecting three RBP trimers at the baseplate periphery. Our goal here was to assess the MTP/Dit interface and the Tal/Dit interface, and to model a complete baseplate structure. To this end, we predicted the structure of a complex of MTP (×6), Dit (×3), and Tal (×3) structural domains (residues 1–380) (3672 residues in total). We also predicted the structure of the full-length Tal trimer and superimposed it onto the MTP/Dit/Tal N-terminal predicted structure using Coot [35] (Figure 8A). Lastly, we predicted the structure of the trimeric BppU, the trimeric RBP, and their complexes, and compared them to the X-ray structures.
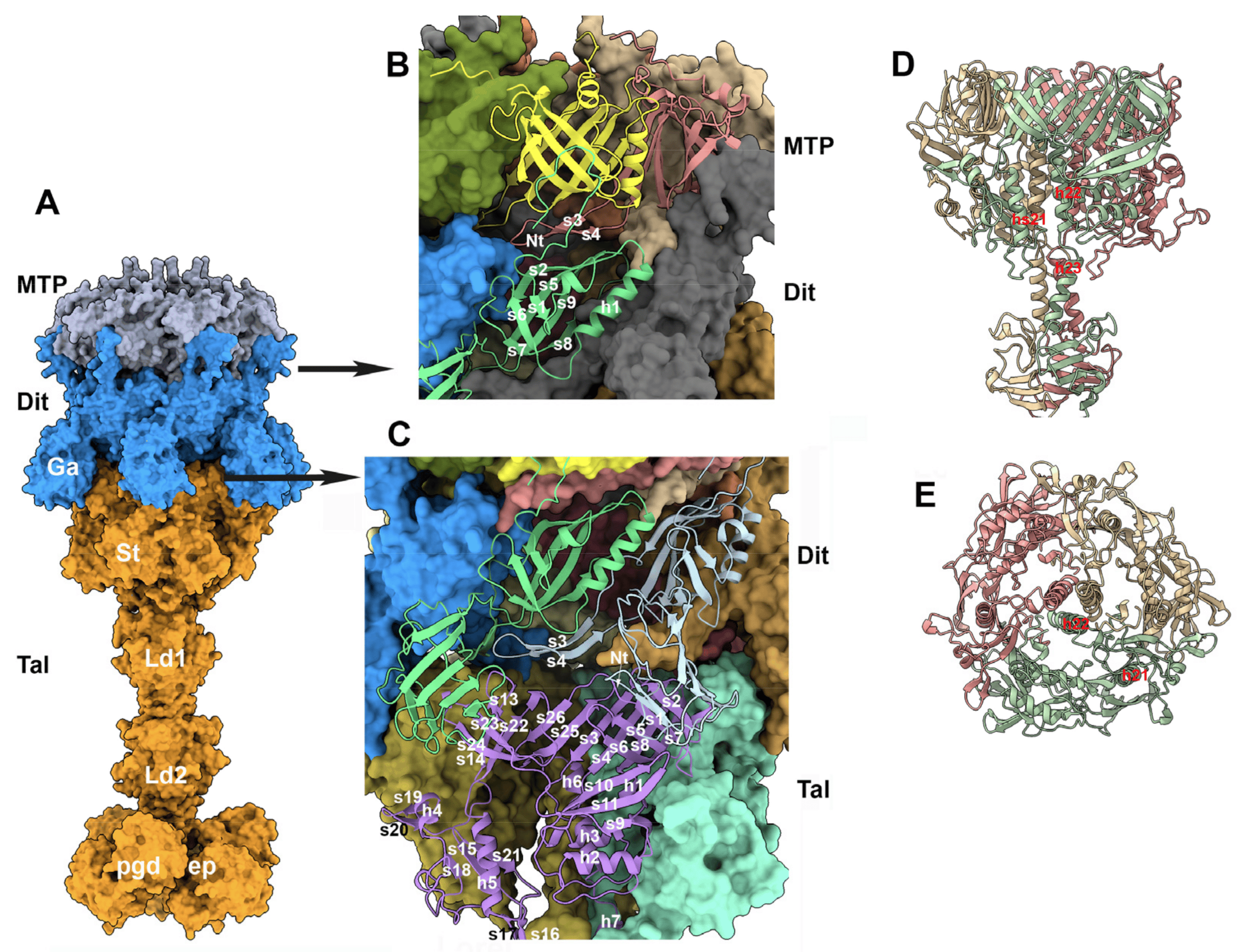
Figure 8. The distal MTP and the central baseplate. (A) Surface view of the distal hexameric MTP (gray), the hexameric Dit (blue), and the trimeric Tal (orange). Ga: the Dit’s galectin domain; St: the Tal’s conserved structural domain; Ld1 and Ld2: linker domains 1 and 2; pgd and endopeptidase (ep) domains. (B) Ribbon view of the MTP/Dit interface. (C) Ribbon view of the Dit/Tal interface. (D) Ribbon side-view of the Tal trimer N-terminus (residues 1–484). (E) Same view rotated by 90° within the phage’s main axis.
4. Discussion
In our predicted structure of the TP901-1 virion, the hexons and pentons of the capsid were well predicted, except for the N-termini, as they may be involved in interactions with other hexons or pentons. Our predictions of hexon/hexon or hexon/penton interactions were unsuccessful: the hexons and penton were predicted as separate units without any interactions. Hence, AF2 cannot determine the contents of an icosahedral asymmetric unit and cannot predict the T-number nor handedness.
The procapsid assembly requires a so-called scaffolding protein (SP), which acts as a chaperone to ensure the correct positioning of the subunits [57,58]. The structures of full or partial SPs have been previously reported (PDB ID: 1tx9, 2gp8, 1no4, 8dt0); they are helical proteins, as seen in the phage Phi29, where they form a dimer of two long a-helices [57]. In the procapsid of staphylococcal phage 80α, the SP C-terminal a-helix was observed to be in contact with each of the capsid components [59]. In phage TP901-1, the predicted structure of the expression product of ORF35, which is located immediately upstream of the MCP-encoding gene, displays a pattern of two long helices and some shorter ones, making it a good candidate for being an SP.
The predicted dodecameric portal exhibits all the characteristics of genuine phage portals previously reported in the literature [45,60,61]. However, the TP901-1 portal has a unique feature: its clip domain contains three β-strands originating from two different portals, and a fourth β-strand coming from the adaptor. The TP901-1 adaptor also presents a unique feature, with a 24-stranded β-barrel forming its internal channel. Interestingly, the not-compact and rather loose interface between the portal and adaptor is compensated by the presence of a ring of six NPS trimers, whose N-terminal domains cover the adaptor ring. The hexameric stopper of TP901-1 is similar to that of other phages and exhibits long b-hairpins that embrace the tail tube upon complexation.
The tail and baseplate components, including the TT, MTP, and Dit proteins, share structural features that have previously been reported [15,62]. A superimposition of their monomers reveals that these three components possess a β-hairpin that interacts with the other subunits of a hexameric ring and also provides a stacking platform between the hexameric rings. The MTP and Dit proteins also possess an extended N-terminus that inserts into a crevice of an above hexamer in the MTP/MTP and Dit/MTP interactions. While platforms of β-hairpins were also observed in the MTP stacking of the tailed phages 80α [48] and T5 [56,63], the N-terminal lock was only observed in 80α, for which a β-helix helps the interaction.
The structure of a TP901-1 baseplate has previously been reported, though without its Tal component [5]. Here, the full-length Tal completes the X-ray structure. The central channel of the Tal N-terminal structural domain (1–380) is filled by three α-helices that link this domain to the functional C-terminal domain. Surprisingly, this central channel is filled in phage 80α by the C-terminus of the three TMPs contained in the tail tube [48]. However, the position of the three helices in TP901-1 Tal is logical considering the Tal topology. Indeed, these helices and the C-terminal domain should dramatically rearrange to allow the TMP to exit and for DNA ejection during the initial stages of infection. Such a rearrangement has recently been reported for phage T5. Upon contact of the tail tip with T5′s receptor (the membrane protein FhuA), the Tal-like protein pb3, which obstructs the tail exit channel, opens and rotates on the tail side, thus allowing the TMP (pb2) to insert into the membrane [10,56]. We postulate that a similar mechanism is operating in the case of phage TP901-1 upon its baseplate/cell wall polysaccharide binding.
Our prediction of the RBP structures and their interaction with BppU was excellent, as was the prediction and localization of 12 out of the 18 BppU N-terminal domains that form a ring similar to the NPS ring or to the tail fiber rings observed in phage 80α [48]. However, the six remaining BppU N-terminal domains and the BppU topology were out of reach due to the fact that the complete structure is well over AF2’s residue limit. Lastly, all attempts to model the TMP in complex with the MTP, Dit, and tail chaperone were unsuccessful.
To conclude, we have found that AF2 has impressive prediction capabilities, provided that the ensemble used for prediction involves a residue limit of less than around 4000. Most of our prediction failures were primarily linked to this limitation. However, as evidenced by the capsid and parts of the baseplate, the limitations of this study may not be due to this factor only. Despite this, we illustrate here that AF2’s predictions provide an avenue for further investigation into phage structures. This is particularly important for phages that remain poorly characterized at the structural level, such as those infecting human pathogens and mycobacteria [64].
References
- Ackermann, H.W. Bacteriophage Electron Microscopy. Adv. Virus Res. 2012, 82, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Adriaenssens, E.M. Abolishment of Morphology-Based Taxa and Change to Binomial Species Names: 2022 Taxonomy Update of the Ictv Bacterial Viruses Subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Tremblay, D.; Moineau, S.; Rattei, T.; Kushkina, A.I.; Tovkach, F.I.; Krisch, H.M.; Ackermann, H.W. Phage Morphology Recapitulates Phylogeny: The Comparative Genomics of a New Group of Myoviruses. PLoS ONE 2012, 7, e40102. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Spinelli, S.; Mahony, J.; Cambillau, C. Conserved and Diverse Traits of Adhesion Devices from Siphoviridae Recognizing Proteinaceous or Saccharidic Receptors. Viruses 2020, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Spinelli, S.; Mahony, J.; Lichiere, J.; Blangy, S.; Bricogne, G.; Legrand, P.; Ortiz-Lombardia, M.; Campanacci, V.; van Sinderen, D.; et al. Structure of the Phage TP901-1 1.8 MDa Baseplate Suggests an Alternative Host Adhesion Mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 8954–8958. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, K.; Goulet, A.; McDonnel, B.; Spinelli, S.; van Sinderen, D.; Mahony, J.; Cambillau, C. Revisiting the Host Adhesion Determinants of Streptococcus Thermophilus Siphophages. Microb. Biotech. 2020, 13, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Tremblay, D.; Farenc, C.; Chapot-Chartier, M.P.; Sadovskaya, I.; van Heel, M.; Veesler, D.; Moineau, S.; Cambillau, C. Structure, Adsorption to Host, and Infection Mechanism of Virulent Lactococcal Phage p2. J. Virol. 2013, 87, 12302–12312. [Google Scholar] [CrossRef]
- Baptista, C.; Santos, M.A.; Sao-Jose, C. Phage SPP1 Reversible Adsorption to Bacillus subtilis cell Wall Teichoic Acids Accelerates Virus Recognition of Membrane Receptor YueB. J. Bacteriol. 2008, 190, 4989–4996. [Google Scholar] [CrossRef]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Chaperone-Protein Interactions That Mediate Assembly of the Bacteriophage Lambda Tail to the Correct Length. J. Mol. Biol. 2014, 426, 1004–1018. [Google Scholar] [CrossRef]
- Degroux, S.; Effantin, G.; Linares, R.; Schoehn, G.; Breyton, C. Deciphering Bacteriophage T5 Host Recognition Mechanism and Infection Trigger. J. Virol. 2023, 97, e0158422. [Google Scholar] [CrossRef]
- Mahony, J.; Stockdale, S.R.; Collins, B.; Spinelli, S.; Douillard, F.P.; Cambillau, C.; van Sinderen, D. Lactococcus lactis phage TP901-1 as a model for Siphoviridae virion assembly. Bacteriophage 2016, 6, e1123795. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Rossmann, M.G. Molecular Architecture of Tailed Double-Stranded DNA Phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Bebeacua, C.; Orlov, I.; Tremblay, D.; Klaholz, B.P.; Moineau, S.; Cambillau, C. Cryo-Electron Microscopy Structure of Lactococcal Siphophage 1358 virion. J. Virol. 2014, 88, 8900–8910. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Joos, R.; Lavelle, K.; van Sinderen, D.; Mahony, J.; Cambillau, C. A structural Discovery Journey of Streptococcal Phages Adhesion Devices by AlphaFold2. Front. Mol. Biosci. 2022, 9, 960325. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Cambillau, C. A Common Evolutionary Origin for Tailed-Bacteriophage Functional Modules and bacterial Machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cruz, S.; Erazo Garzon, A.; Kelleher, P.; Bottacini, F.; Breum, S.O.; Neve, H.; Heller, K.J.; Vogensen, F.K.; Palussiere, S.; Courtin, P.; et al. Host Genetic Requirements for DNA Release of Lactococcal Phage TP901-1. Microb. Biotechnol. 2022, 15, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, S.R.; Collins, B.; Spinelli, S.; Douillard, F.P.; Mahony, J.; Cambillau, C.; van Sinderen, D. Structure and Assembly of TP901-1 Virion Unveiled by Mutagenesis. PLoS ONE 2015, 10, e0131676. [Google Scholar] [CrossRef]
- Ostergaard Breum, S.; Neve, H.; Heller, K.J.; Vogensen, F.K. Temperate Phages TP901-1 and phiLC3, Belonging to the P335 Species, Apparently Use Different Pathways for DNA Injection in Lactococcus lactis subsp. cremoris 3107. FEMS Microbiol. Lett. 2007, 276, 156–164. [Google Scholar] [CrossRef]
- Rasmussen, K.K.; Palencia, A.; Varming, A.K.; El-Wali, H.; Boeri Erba, E.; Blackledge, M.; Hammer, K.; Herrmann, T.; Kilstrup, M.; Lo Leggio, L.; et al. Revealing the Mechanism of Repressor Inactivation during Switching of a Temperate Bacteriophage. Proc. Natl. Acad. Sci. USA 2020, 117, 20576–20585. [Google Scholar] [CrossRef]
- Pedersen, M.; Neergaard, J.T.; Cassias, J.; Rasmussen, K.K.; Lo Leggio, L.; Sneppen, K.; Hammer, K.; Kilstrup, M. Repression of the lysogenic P(R) Promoter in Bacteriophage TP901-1 through Binding of a CI-MOR Complex to a Composite O(M)-O(R) Operator. Sci. Rep. 2020, 10, 8659. [Google Scholar] [CrossRef]
- Varming, A.K.; Rasmussen, K.K.; Zong, Z.; Thulstrup, P.W.; Kilstrup, M.; Lo Leggio, L. Flexible linker modulates the binding affinity of the TP901-1 CI phage repressor to DNA. FEBS J. 2022, 289, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Lai, L.; Vegge, C.S.; Brondsted, L.; van Heel, M.; Veesler, D.; Cambillau, C. Visualizing a Complete Siphoviridae Member by Single-Particle Electron Microscopy: The Structure of Lactococcal Phage TP901-1. J. Virol. 2013, 87, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Vegge, C.S.; Brondsted, L.; Neve, H.; Mc Grath, S.; van Sinderen, D.; Vogensen, F.K. Structural characterization and assembly of the distal tail structure of the temperate lactococcal bacteriophage TP901-1. J. Bacteriol. 2005, 187, 4187–4197. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, S.; Sadovskaya, I.; Vinogradov, E.; Courtin, P.; Guerardel, Y.; Mahony, J.; Grard, T.; Cambillau, C.; Chapot-Chartier, M.P.; van Sinderen, D. Differences in Lactococcal Cell Wall Polysaccharide Structure Are Major Determining Factors in Bacteriophage Sensitivity. MBio 2014, 5, e00880-14. [Google Scholar] [CrossRef] [PubMed]
- Sciara, G.; Bebeacua, C.; Bron, P.; Tremblay, D.; Ortiz-Lombardia, M.; Lichiere, J.; van Heel, M.; Campanacci, V.; Moineau, S.; Cambillau, C. Structure of Lactococcal Phage p2 Baseplate and Its Mechanism of Activation. Proc. Natl. Acad. Sci. USA 2010, 107, 6852–6857. [Google Scholar] [CrossRef] [PubMed]
- Mahony, J.; Alqarni, M.; Stockdale, S.; Spinelli, S.; Feyereisen, M.; Cambillau, C.; Sinderen, D.V. Functional and Structural Dissection of the Tape Measure Protein of Lactococcal Phage TP901-1. Sci. Rep. 2016, 6, 36667. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and Improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Jumper, J.; Hassabis, D. Protein Structure Predictions to Atomic Accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Holm, L.; Kaariainen, S.; Rosenstrom, P.; Schenkel, A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Choi, K.H.; Koti, J.S.; Chipman, P.R.; Anderson, D.L.; Rossmann, M.G. Conservation of the Capsid Structure in tailed dsDNA Bacteriophages: The Pseudoatomic Structure of phi29. Mol. Cell 2005, 18, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Helgstrand, C.; Wikoff, W.R.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E.; Liljas, L. The Refined Structure of a Protein Catenane: The HK97 Bacteriophage Capsid at 3.44 A Resolution. J. Mol. Biol. 2003, 334, 885–899. [Google Scholar] [CrossRef]
- Rao, V.B.; Fokine, A.; Fang, Q.; Shao, Q. Bacteriophage T4 Head: Structure, Assembly, and Genome Packaging. Viruses 2023, 15, 527. [Google Scholar] [CrossRef]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Wall, E.A.; Klenow, L.; Hill, R.L.L.; Spilman, M.S.; Stagg, S.M.; Christie, G.E.; Dokland, T. Cleavage and Structural Transitions during Maturation of Staphylococcus aureus Bacteriophage 80α and SaPI1 Capsids. Viruses 2017, 9, 384. [Google Scholar] [CrossRef]
- Ignatiou, A.; Brasiles, S.; El Sadek Fadel, M.; Burger, J.; Mielke, T.; Topf, M.; Tavares, P.; Orlova, E.V. Structural Transitions during the Scaffolding-Driven Assembly of a Viral Capsid. Nat. Commun. 2019, 10, 4840. [Google Scholar] [CrossRef]
- Lebedev, A.A.; Krause, M.H.; Isidro, A.L.; Vagin, A.A.; Orlova, E.V.; Turner, J.; Dodson, E.J.; Tavares, P.; Antson, A.A. Structural Framework for DNA Translocation via the Viral Portal Protein. EMBO J. 2007, 26, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Fokine, A.; Fang, Q. The Remarkable Viral Portal Vertex: Structure and a Plausible Model for Mechanism. Curr. Opin. Virol. 2021, 51, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Feiss, M.; Rao, V.B. The Bacteriophage DNA Packaging Machine. Adv. Exp. Med. Biol. 2012, 726, 489–509. [Google Scholar] [CrossRef] [PubMed]
- Orlov, I.; Roche, S.; Brasiles, S.; Lukoyanova, N.; Vaney, M.C.; Tavares, P.; Orlova, E.V. CryoEM Structure and Assembly Mechanism of a Bacterial Virus Genome Gatekeeper. Nat. Commun. 2022, 13, 7283. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fabrega-Ferrer, M.; Machon, C.; Conesa, J.J.; Fernandez, F.J.; Perez-Luque, R.; Perez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 Bacteriophage Portal and Tail Suggest a Viral DNA Retention and Ejection Mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed]
- Bardy, P.; Fuzik, T.; Hrebik, D.; Pantucek, R.; Thomas Beatty, J.; Plevka, P. Structure and Mechanism of DNA Delivery of a Gene Transfer Agent. Nat. Commun. 2020, 11, 3034. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Mahony, J.; Cambillau, C.; van Sinderen, D. Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2. Microorganisms 2022, 10, 2278. [Google Scholar] [CrossRef]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Dokland, T. Structure of the Host Cell Recognition and Penetration Machinery of a Staphylococcus aureus Bacteriophage. PLoS Pathog. 2020, 16, e1008314. [Google Scholar] [CrossRef]
- Zinke, M.; Sachowsky, K.A.A.; Oster, C.; Zinn-Justin, S.; Ravelli, R.; Schroder, G.F.; Habeck, M.; Lange, A. Architecture of the Flexible Tail Tube of Bacteriophage SPP1. Nat. Commun. 2020, 11, 5759. [Google Scholar] [CrossRef]
- Labrie, S.J.; Josephsen, J.; Neve, H.; Vogensen, F.K.; Moineau, S. Morphology, Genome Sequence, and Structural Proteome of type Phage P335 from Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74, 4636–4644. [Google Scholar] [CrossRef]
- Mahony, J.; Oliveira, J.; Collins, B.; Haanemaaijer, L.; Lugli, G.A.; Neve, H.; Ventura, M.; Kouwen, T.R.; Cambillau, C.; van Sinderen, D.; et al. Genetic and Functional Characterisation of the Lactococcal P335 Phage-Host Interactions. BMC Genom. 2017, 18, 146. [Google Scholar] [CrossRef]
- Veesler, D.; Robin, G.; Lichiere, J.; Auzat, I.; Tavares, P.; Bron, P.; Campanacci, V.; Cambillau, C. Crystal Structure of Bacteriophage SPP1 Distal Tail Protein (gp19.1): A Baseplate Hub Paradigm in Gram-Positive Infecting Phages. J. Biol. Chem. 2010, 285, 36666–36673. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the Cell-Puncturing Device of Bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 Baseplate and Its Function in Triggering Sheath Contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef]
- Stockdale, S.R.; Mahony, J.; Courtin, P.; Chapot-Chartier, M.P.; van Pijkeren, J.P.; Britton, R.A.; Neve, H.; Heller, K.J.; Aideh, B.; Vogensen, F.K.; et al. The lactococcal phages Tuc2009 and TP901-1 Incorporate Two Alternate Forms of Their Tail Fiber into Their Virions for Infection Specialization. J. Biol. Chem. 2013, 288, 5581–5590. [Google Scholar] [CrossRef] [PubMed]
- Linares, R.; Arnaud, C.A.; Effantin, G.; Darnault, C.; Epalle, N.H.; Boeri Erba, E.; Schoehn, G.; Breyton, C. Structural Basis of Bacteriophage T5 Infection Trigger and E. coli Cell Wall Perforation. Sci. Adv. 2023, 9, eade9674. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Kanamaru, S.; Badasso, M.O.; Koti, J.S.; Owen, B.A.; McMurray, C.T.; Anderson, D.L.; Rossmann, M.G. BACTERIOPHAGE phi29 Scaffolding Protein gp7 before and after Prohead Assembly. Nat. Struct. Biol. 2003, 10, 572–576. [Google Scholar] [CrossRef]
- Li, S.; Roy, P.; Travesset, A.; Zandi, R. Why Large Icosahedral Viruses Need Scaffolding Proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10971–10976. [Google Scholar] [CrossRef]
- Dearborn, A.D.; Wall, E.A.; Kizziah, J.L.; Klenow, L.; Parker, L.K.; Manning, K.A.; Spilman, M.S.; Spear, J.M.; Christie, G.E.; Dokland, T. Competing Scaffolding Proteins Determine Capsid Size during Mobilization of Staphylococcus aureus Pathogenicity Islands. eLife 2017, 6, e30822. [Google Scholar] [CrossRef]
- Orlova, E.V.; Dube, P.; Beckmann, E.; Zemlin, F.; Lurz, R.; Trautner, T.A.; Tavares, P.; van Heel, M. Structure of the 13-Fold Symmetric Portal Protein of Bacteriophage SPP1. Nat. Struct. Biol. 1999, 6, 842–846. [Google Scholar]
- Cuervo, A.; Vaney, M.C.; Antson, A.A.; Tavares, P.; Oliveira, L. Structural Rearrangements between Portal Protein Subunits Are Essential for Viral DNA Translocation. J. Biol. Chem. 2007, 282, 18907–18913. [Google Scholar] [CrossRef]
- Linares, R.; Arnaud, C.A.; Degroux, S.; Schoehn, G.; Breyton, C. Structure, Function and Assembly of the Long, Flexible Tail of Siphophages. Curr. Opin. Virol. 2020, 45, 34–42. [Google Scholar] [CrossRef]
- Arnaud, C.A.; Effantin, G.; Vives, C.; Engilberge, S.; Bacia, M.; Boulanger, P.; Girard, E.; Schoehn, G.; Breyton, C. Bacteriophage T5 Tail Tube Structure Suggests a Trigger Mechanism for Siphoviridae DNA ejection. Nat. Commun. 2017, 8, 1953. [Google Scholar] [CrossRef]
- Cambillau, C.; Goulet, A. Exploring Host-Binding Machineries of Mycobacteriophages with AlphaFold2. J. Virol. 2023, 97, e0179322. [Google Scholar] [CrossRef]
