


1. Introduction
Breast cancer (BC) has the highest global incidence in women [1]. Up to 16% of women worldwide die every year from causes related to this disease [2]. Similarly to other types of cancer, BC is a genetic disease characterized by the presence of genomic alterations [3] that can be either inherited or acquired throughout life [4]. Only a reduced set of genomic alterations identified so far trigger cellular transformations that lead to the onset of cancer and tumor growth [5,6]. However, many genes still require thorough analysis to unravel their potential key role in tumor development and establishing high-risk cancer phenotypes.
ARID4B, also known as RBBP1-like protein, is a member of the ARID (AT-rich Interaction Domain) family, which modulates the activity of genes involved in cell proliferation and chromatin remodeling [7]. The Human Protein Atlas and previous studies of ARID4B in normal tissues show transcriptional levels in the testes, brain, skin, gallbladder, and, to a minor extent, the thymus, prostate, and ovary [8,9]. In addition, ARID4B and ARID4A have been found to regulate PWS/AS (Prader–Willi syndrome and Angelman syndrome), indicating that they may play an important role in epigenetic mechanisms [10].
We noted from a previous study that ARID4B mutations are related to a reduced survival time [11] and that other members of the ARID family are well-studied in BC. For example, ARID1A and ARID1B are known to be driver genes in BC [12], while mutations in estrogen receptor-positive BC show a higher risk [13]. Moreover, low expression of ARID1A is associated with a more aggressive phenotype [14]. The high expression of ARID1B is also associated with a higher histological grade and tumor size [15]. In addition, the silencing of this gene in triple-negative cells produces a delay in the cell cycle [15], indicating that different members of the ARID family could be associated with better or worse prognosis in cancer, suggesting that differences in domains and protein sequences lead to different outcomes. Thus, we focus on ARID4B, whose mutations have not been studied deeply.
Transcriptional upregulation of ARID4B has been detected in various human carcinomas. In hepatocellular carcinoma, levels of ARID4B show a positive correlation with tumor progression and adverse prognosis [16]. In prostate cancer, especially in the PTEN-null phenotype, upregulation of ARID4B activates the PTEN-PI3K pathway through specific transactivation of both PIK3CA and PIK3R2, which favors tumor progression and correlates positively with prostate cancer recurrence [17]. Contrary, in human leukemias, ARID4B seems to exert a tumor suppressor activity, and either its downregulation or heterozygous inactivating mutations alongside mutations in ARID4A in vivo lead to leukemic transformation through indirect epigenetic mechanisms [18]. In mice, ARID4B expression has been found to promote the proliferation and invasiveness of malignant cells; in fact, expression increases tumor growth in vivo and in vitro [19]. In addition, variations in miR-290, which targets ARID4B, have been validated in in vitro models, reducing proliferation and metastasis [20]. Using databases of cancer-related hotspots, we have found that the ARID4B gene has two hotspots that have never been validated [21].
Overall, the above evidence shows that ARID4B has an important role in BC and observed recurrent mutations of ARID4B in BC patients suggest a possible impact. However, there are no previous analyses about the effects of ARID4B loss-of-function in human cells. In this work, we generated loss-of-function mutations by targeting ARID4B in MCF-7 cells using the CRISPR/Cas9 system in one of these critical hotspots previously found. After validation by targeted and exome sequencing, we evaluated how the disruption of ARID4B impacts the viability and functionality of BC cells by analyzing cell proliferation, viability, and migration. Furthermore, an expression analysis using RNA-seq in MCF-7 cells with loss-of-function mutations in ARID4B compared to the wild-type shows the transcriptional effect of these mutations. Our results shed light on the importance of ARID4B mutations in cancer.
2. Materials and Methods
2.1. Expression and Survival Analysis
Information related to expression levels of ARID4B in breast cancer and normal human tissues was obtained from TCGA (The Cancer Genome Atlas) data using the Gene Expression Profiling Interactive Analysis (GEPIA) platform [22], available at http://gepia.cancer-pku.cn/detail.php?gene=ARID4B (accessed on 21 October 2022). TCGA information about survival analyses of breast cancer patients and normal human tissues was obtained using The University of Alabama at Birmingham Cancer Data Analysis Portal (UALCAN) [23], available at http://ualcan.path.uab.edu (accessed on 7 February 2023). Analysis of the effects exerted by mutations in ARID4B on the survival of BC patients was carried out on TCGA data using the tool VALORATE (Velocity and Accuracy of the Log-Rank Test) [11]. All figures obtained from these platforms were adapted for this manuscript. Identification of recurrent mutations in the ARID4B sequence was carried out using the HotspotAnnotations Database [21].
2.2. Cell Culture and Transfection
Protocols for culture and transfection of MCF-7 cells have been described previously [24]. Briefly, MCF-7 cells were cultured at 37 °C, 95% humidity, and 5% CO2 in a culture medium containing DMEM-F/12 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.). Cells were cultured at 70% of confluence in 6-well plates and were then transfected with 4 µg of pX459 vector or pX459-ARID4B construct using 6 µL of XtremeGene9 Transfection Reagent (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and 200 µL of Optimem Medium (Gibco; Thermo Fisher Scientific, Inc.). Plasmid pX459 was a gift from Feng Zhang (Addgene plasmid # 62988; http://n2t.net/addgene:62988; RRID:Addgene_62988) [25]. After 48 h, medium was replaced, and fresh medium with 6 µg/mL of puromycin (Sigma-Aldrich; Merck KGaA) was added. Again, after 48 h, when cells in the controls were all dead, the selection was finished, and fresh medium was added to recover and grow the cells for one week.
2.3. sgRNA Design and CRISPR/Cas9 System Construction
sgRNA sequences for CRISPR/Cas9 were designed using the CRISPR design tool (previously available at http://crispr.mit.edu/ (accessed on 23 June 2017) provided by the Feng Zhang Lab. These sgRNA targets near the amino acid 939 in the protein Arid4b, an important hotspot found in HotspotAnnotations Data Base. The sequences of this sgRNA are 5′-CACCGCCAGACATCTTTTCGATCTT-3′ (top) and 5′-AAACAAGAT CGAAAAGATGTCTGGC-3′ (bottom). Both oligos were aligned and cloned in Bbs1 digested pX459 (62988, Addgene, Cambridge, MA, USA). Then, we expanded the pX459-ARID4B vector using calcium-competent Escherichia coli DH5α. Correct insertion of sgRNA into the pX459 vector was confirmed using Sanger sequencing.
2.4. Isolation of Transfected Clones
Single-cell derived colonies were obtained through limiting dilution of cell pools that had been previously transfected with the pX459-ARID4B vector and enriched upon exposure to puromycin. For this, confluent cell pools were trypsinized and counted using Trypan blue (Gibco; Thermo Fisher Scientific, Inc.). Cell suspensions were serially diluted using DMEM/F12, and cell density was adjusted to 5 cells/mL. A total of 100 µL of this solution was transferred to each well of a 96-well plate, and single cells were incubated under standard conditions. Then, 3 days after cell seeding, 96-well plates were inspected through the microscope, and wells containing single cells were selected for further experimentation. Cells were genotyped by Surveyor assay and Sanger sequencing. One of the clones that resulted negative for genetic editions in ARID4B was used as negative control (NC).
2.5. Validation of Loss-of-Function Mutations
Isolation of DNA from transfected monoclonal cells was carried out using the GenEluteTM Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich; Merck KGaA). Amplification of an 838 bp region of ARID4B was performed by PCR, using the GoTaq® DNA Polymerase (Promega Corporation, Madison, WI, USA) with the following primers: 5′- GCTGAAGACAGTGAGCAGGA-3′and 5′-CGACATTGACTGGAGGTGGT-3′. The presence of mutations in the amplified genomic region was confirmed through the Surveyor assay. For this, PCR amplicons were denatured and reannealed for heteroduplex formation. DNA heteroduplexes were processed using the Surveyor® Mutation Detection Kit (IDT, Coralville, IA, USA). DNA fragments obtained upon enzyme-mediated detection of mismatching nucleotides and cleavage of DNA sequences were separated through agarose gel electrophoresis. Clones carrying the correct genomic modification at ARID4B were detected through Sanger sequencing using the BigDye Terminator v1.1 Cycle Sequencing Kit (Invitrogen, Thermo Fisher Scientific, Inc.) as per manufacturer’s instructions. Briefly, a master mix containing 1 µL of Big Dye Terminator and 2 µL of Buffer 5X was mixed with 3.2 pmol of primer 5′-GCTGAAGAGAGTGAGCAGGA-3′ and 20 ng of purified PCR product. Then, the reaction volume was adjusted to 10 µL using nuclease-free water. Amplified products were purified using the BigDye X-Terminator Purification Kit (Invitrogen, Thermo Fisher Scientific, Inc.) and analyzed using the ABI PRISM 3100 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Waltham, MA, USA). Sequences were analyzed using the INDIGO platform [26] at https://gear.embl.de/indigo/ (accessed on 28 October 2018) to detect mutations.
2.6. Whole Exome Sequencing and Analysis of Off-Target Effects
Whole-Exome Sequencing (WES) was carried out as follows. First, DNA was isolated from cells as described previously. Isolated DNA was quantified using the Qubit™ dsDNA H.S. Assay Kit (Invitrogen, Thermo Fisher Scientific, Inc.) and the Qubit 4 fluorometer. Exome sequencing was performed using the Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA, USA) via a paired-end for 100-bp protocol following the manufacturer’s instructions. Reads were mapped to the reference human genome (hg38) using the BWA-MEM alignment algorithm [27] and visualized using the Integrative Genomics Viewer (IGV) program [28]. Post-processing of data was carried out using the Galaxy web platform [29]. Potential off-target effects of the selected sgRNAs were analyzed using the CRISPR-Cas9 guide RNA design checker developed by IDT, available at https://www.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE (accessed on 23 January 2023); and genes with a higher score for potential off-target effects (ZNF615, TOP2B, LACTB, and LDLRAP1) were analyzed through WES. BioProject accesion number in NCBI of WES data is PRJNA930489 https://www.ncbi.nlm.nih.gov/sra/PRJNA930489.
2.7. Expression Analysis
Total RNA was isolated from clones using GenElute™ Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich; Merck KGaA). The integrity of RNA was analyzed through gel electrophoresis. cDNA was synthesized from 1 µg of purified RNA using the High-Capacity cDNA Reverse Transcription Kit (Invitrogen; Thermo Fisher Scientific, Inc.). qPCR reactions were prepared using the PowerUp™ SYBR® Green Master Mix (Invitrogen; Thermo Fisher Scientific, Inc.) and primers: 5′-GGCCCACTAAAGGTAGGAGC-3′ and 5′-AGTGTACCAACTCGCATCTGT-3′). GAPDH was used as a housekeeping gene to normalize Ct values, using primers: 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-TTGATTTTGGAGGGATCTCG-3′). The relative expression of each amplicon was analyzed by the 2−ΔΔCt method [30].
2.8. Cell Growth Analysis
A total of 2 × 103 cells were seeded in each well of 96-well plates containing 90 µL of supplemented DMEM/F12. Following 24 h of incubation, 10 µL of Alamar Blue reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was added to each well, and cells were incubated for 4 h. Then, the fluorescence signal was acquired at 530 nm and 590 nm for excitation and emission wavelengths, respectively. Cell proliferation was measured at 24, 48, and 72 h, using CyQUANT® Direct Cell Proliferation Assay (Invitrogen; Thermo Fisher Scientific, Inc.), following the manufacturer’s instructions. In both cases, samples were read using a Bio-Tek Synergy HT Multi-Detection Microplate Reader (Bio Tek Instruments, Inc., Winooski, VT, USA).
2.9. Cell Migration Assay
The migration of cells was tested using the in vitro scratch assay [31]. Briefly, 24-well plates were seeded with 2.4 × 105 cells per well in triplicates. After overnight incubation under standard conditions, the attachment of cells was confirmed through observation under the microscope. Then, a region of attached cells was cleared in each well through manual scratch, using a 200 µL pipet tip, washing with PBS, and adding fresh culture medium. The area corresponding to the induced wound was identified through the microscope and marked as a reference for future measurements. Plates were incubated for 24 h. Then, the area corresponding to the induced scratch (i.e., the wound) was photographed under a light microscope and analyzed using Image J. Cell migration was determined as the percentage of the area corresponding to the induced wound that had been covered by migrating cells after 24 h of incubation.
2.10. RNA-seq and Enrichment Analysis
RNA was isolated from the clones carrying genomic modifications at ARID4B and negative control cells (NC). RNA integrity was verified using agarose gel and a Nanodrop instrument. RNA libraries were prepared using 300 ng of total RNA and the TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA, USA). The quality of the libraries was verified by the Qubit fluorescence instrument and protocols. Sequencing was carried out using a MiSeq Reagent Kit v3 in a MiSeq Sequencer (Illumina, San Diego, CA, USA), as instructed by manufacturer. A total of 14 million reads passing filter, 76 × 2 cycles, pair-end, 91% Q30, and 50–52% GC content were obtained for the 4 cell types (3 edited clones and negative control), averaging 3.6 M reads per cell type and a minimum of 2.9 M reads. Processing of RNA-seq data was carried out using the Galaxy web platform [32]. Quality control was performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were trimmed using TRIMMOMATIC [33] and mapped using HiSAT2 [34] using hg38 as the human reference genome and default parameters. Read counts were then obtained with FeatureCounts [35]. Because genetic editions are minor, differences in gene expression were expected to be subtle; for that reason, differential gene expression was defined as a 1-fold difference (fold-change defined as log2(A/B)) in samples that had more than 20 reads, following library size (read count) normalization and logarithm transformation processed in the R language (https://cran.r-project.org/). Transcriptomic data are available in GEO at NCBI with the accession number GSE224514 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224514. Functional enrichment analysis was carried out using ENRICH R [36], considering only functional terms with a Benjamini–Hochberg FDR-adjusted-p-value (q-value) < 0.1 (10% FDR) and overlapping with more than three genes.
2.11. Statistical Analysis
Data were analyzed using GraphPad and Prism 5. Student’s t-test was used to assess differences. A value of p < 0.05 was considered statistically significant in all the experiments. When apply * indicates p < 0.05, ** indicates p < 0.005, *** indicates p < 0.0005. Principal Components Analysis (PCA) was performed in the R language using the prcomp function.
3. Results
3.1. ARID4B Expression and Mutations Are Associated with Survival in BC Patients
To know the effect of ARID4B expression in BC patients, we analyzed data from TCGA and the GEPIA database [22] and compared the expression of ARID4B between BC patients and normal individuals. The mean ARID4B expression level was not statistically different across groups (Figure 1A). However, BC patients who had higher levels of ARID4B expression showed lower survival times compared to those BC patients who had low/mid expression levels of ARID4B (Figure 1B). Additionally, we identified the different mutations across the sequence of ARID4B to know the survival probabilities of BC patients. Although the group of mutated patients is tiny (n = 10), our analysis revealed that patients carrying mutations in the ARID4B sequence had significantly lower survival rates compared to patients bearing no mutations in that locus (Figure 1C). To better understand the effect of loss-of-function mutations in the ARID4B gene, we selected the most significant hotspot at the amino acidic position 939 for validation assays according to HotSpotAnnotations (Figure 1D). Although we chose this specific mutation, the effects of frameshifts generated by CRISPR/Cas9 would be similar to those losses of function mutations in DNA positions of T939 and further.
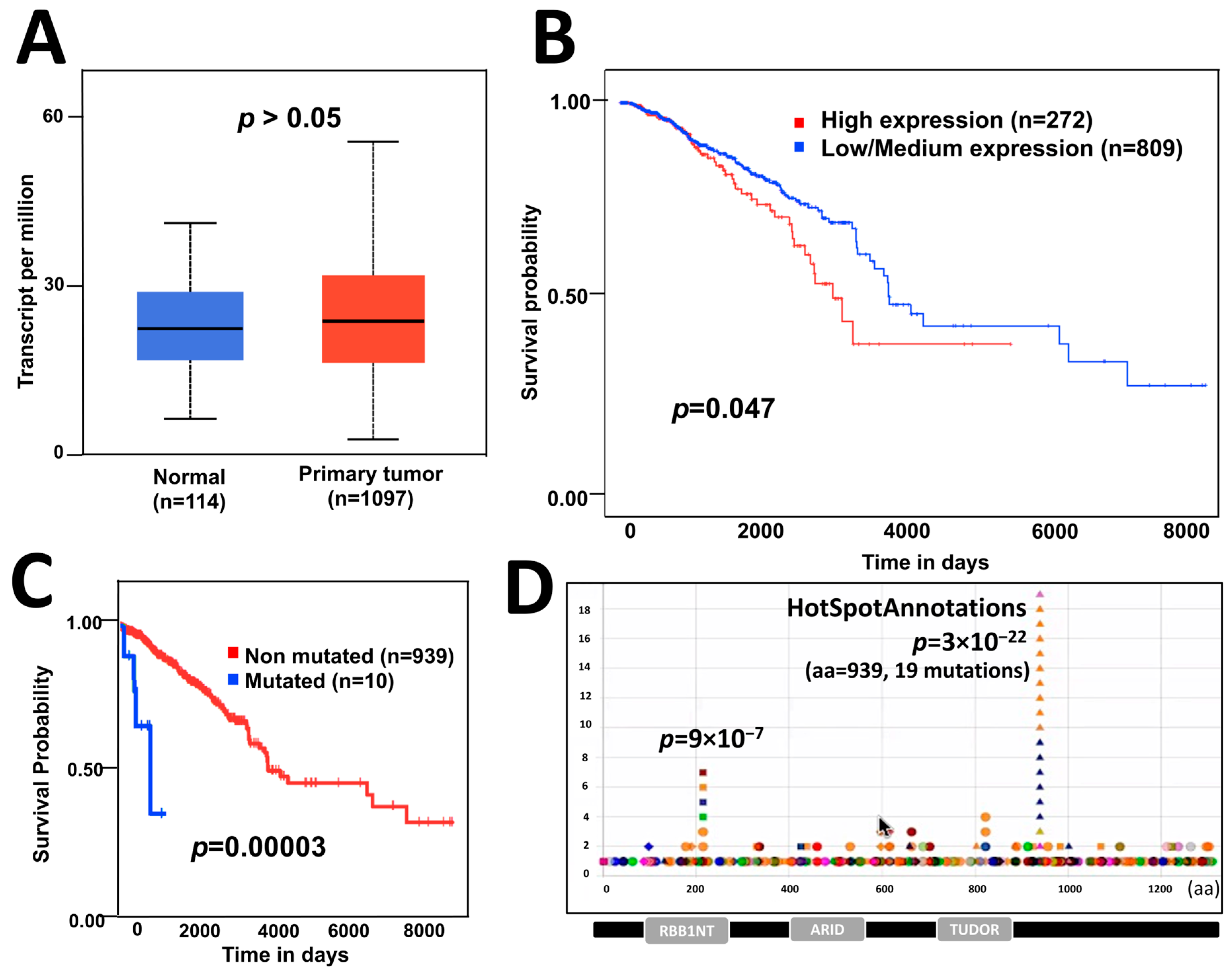
Figure 1. The effect of ARID4B expression and mutations on patient survival. (A) Expression of ARID4B is similar in patients with breast cancer compared with normal tissue, from GEPIA data. (B) High expression of ARID4B is associated with poor survival in patients with breast cancer from TCGA data. (C) Point mutations in ARID4B are associated with low survival in breast cancer patients using statistical analysis for unbalanced groups (VALORATE). (D) Hotspot found in the HotspotAnnotations database to be analyzed in further experiments.
3.2. Generation of ARID4B Knock-Out in the MCF-7 Cell Line
We focus on editing the region of the ARID4B hotspot at position 939 (Figure 2A) by using a region-specific sgRNA and the CRISPR/Cas9 system cloned into the pX459 vector. The sequence of the ligated vector was verified by Sanger sequencing (Figure 2B) and transfected into MCF-7 cells. The cell pool was verified using the Surveyor assay by digesting the 838 bp PCR product from the ARID4B region. After digestion, the two expected fragments of approximately 628 bp and 210 bp were clearly observed, indicating the edition of the target region (Figure 2C). After the selection of seven monoclonal populations by limiting dilution, three clones carrying the genetic edition of the ARID4B region gene were obtained (Figure 2D). From here, these three clones will be named ARID4B-3, ARID4B-10, and ARID4B-21.
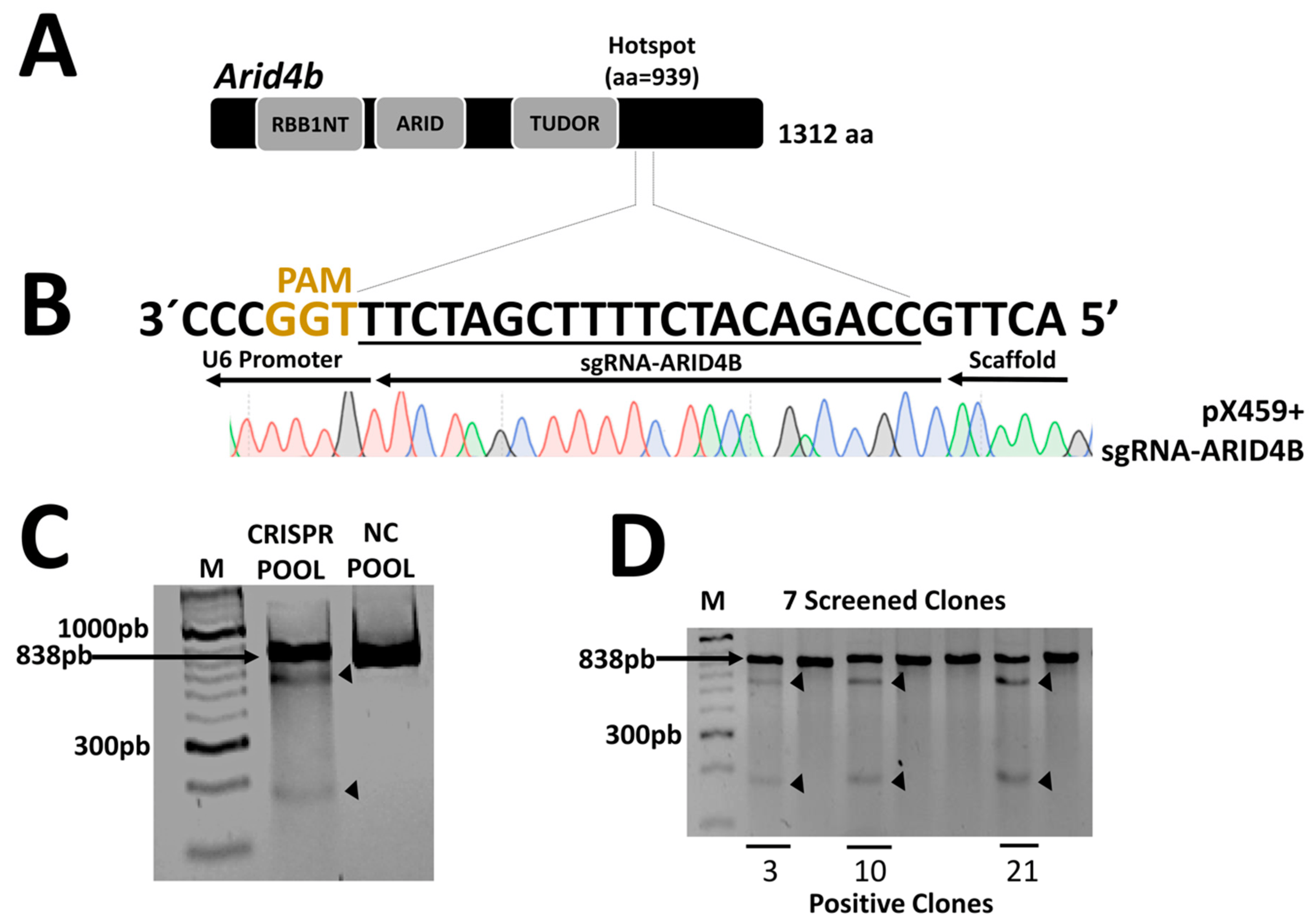
Figure 2. Construction and validation of the CRISPR/Cas9 system. (A) The structure of the ARID4B protein with a total of 1312 amino acids and the site where the mutations were generated in position 939, indicated as a hotspot. (B) Design of the sgRNA and validation of the ligation in the pX459 vector by Sanger sequencing. Only underlined region is shown. Red for T, Black for G, Blue for C, and Green for A. (C) Confirmation of the presence of genetic editions in the pool of MCF-7 cells after transfection. The surveyor assay produces a cut in the PCR product, generating two fragments indicated by arrows. (D) Validation of the presence of genetic editions in isolated clones. Here, 3, 10, and 21 represent the clones positive for mutations in ARID4B.
3.3. Characterization of Genetic Editions in ARID4B-Edited MCF-7 Clones
To identify the precise genetic edition obtained in the three clones, the region flanking the site of interest in this gene was sequenced using Sanger sequencing (Figure 3A). Electropherograms were characterized using the INDIGO platform to determine the specific mutations. Then, using the reads from the whole exome sequencing assay, we noted that the site of interest was indeed correctly edited as described, as shown in the selected examples (Figure 3B). As a result, it was found that clones ARID4B-3 and ARID4B-21 had the same heterozygous insertion of adenine that may produce a truncated protein of 936 amino acids compared to the wild-type protein, which has 1312 amino acids. On the other hand, the clone ARID4B-10 carries a deletion that also affects the reading frame of this gene, which would produce a truncated protein of 939 amino acids (Figure 3C). This confirms the correct introduction of heterozygous loss-of-function mutations in the three clones producing altered versions of the original protein. We performed WES experiments to evaluate the target and off-target regions further. We prioritized regions listed by the software used (see Section 2), which revealed four genes (ZNF615, TOP2B, LACTB, LDLRAP1, see Figure 4). The off-target region for two of these genes are exons (TOP2B and LACTB) that can be analyzed by our exome sequencing experiment. We did not find any recognized edition around the expected off-target site for these two genes relative to wild-type in the cell line, confirming that the edition was specific for the target site in the ARID4B gene (Figure 4). By RNA-Seq, we also confirmed that the alleles carrying genetic editions were indeed transcribed in ARID4B-10 and ARID4B-21 clones (Figure 3D). In the ARID4B-3 clone, we only observed one read of the unedited allele (due to the low number of reads sequenced in that clone). Therefore, the possible functional effects should be attributed to ARID4B editions.
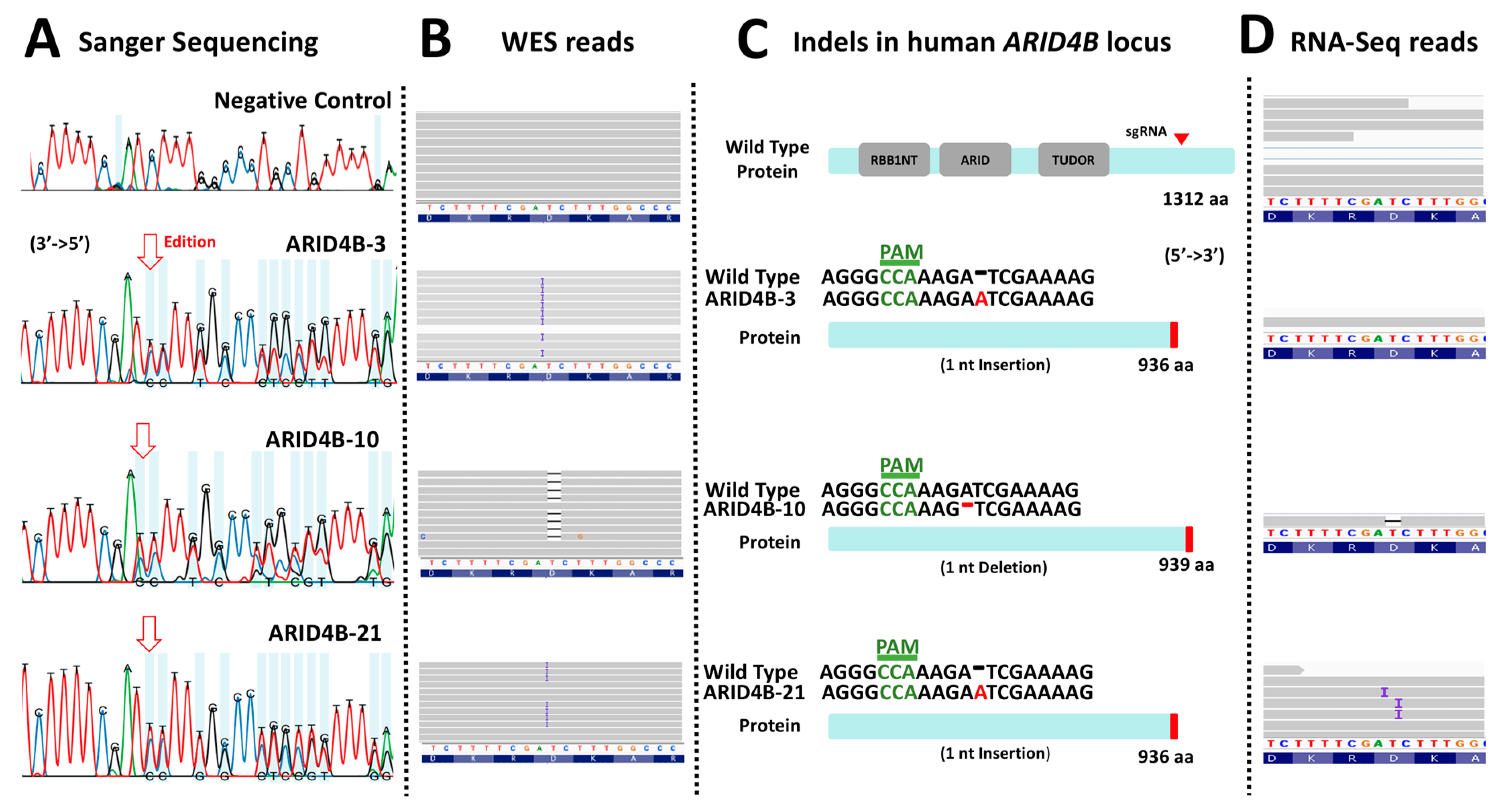
Figure 3. Characterization of loss-of-function mutations in ARID4B. (A) The result of the Sanger sequencing showing altered electropherograms in the three clones previously validated by Surveyor assay. (B) Result of the WES analysis of the three clones. Clones ARID4B-3 and ARID4B-21 have an adenine insertion. On the other hand, clone ARID4B-10 has an adenine deletion. (C) Characterization of the effect of the mutations found in the three clones. Two of the clones (ARID4B-3 and ARID4B-21) alter the reading frame, producing a hypothetical truncated protein of 936 amino acids. Clone ARID4B-10 would produce a 939 amino acid truncated protein. (D) RNA-Seq reads showing that edited DNA have been transcribed in at least two clones (ARID4B-10 and ARID4B-21).
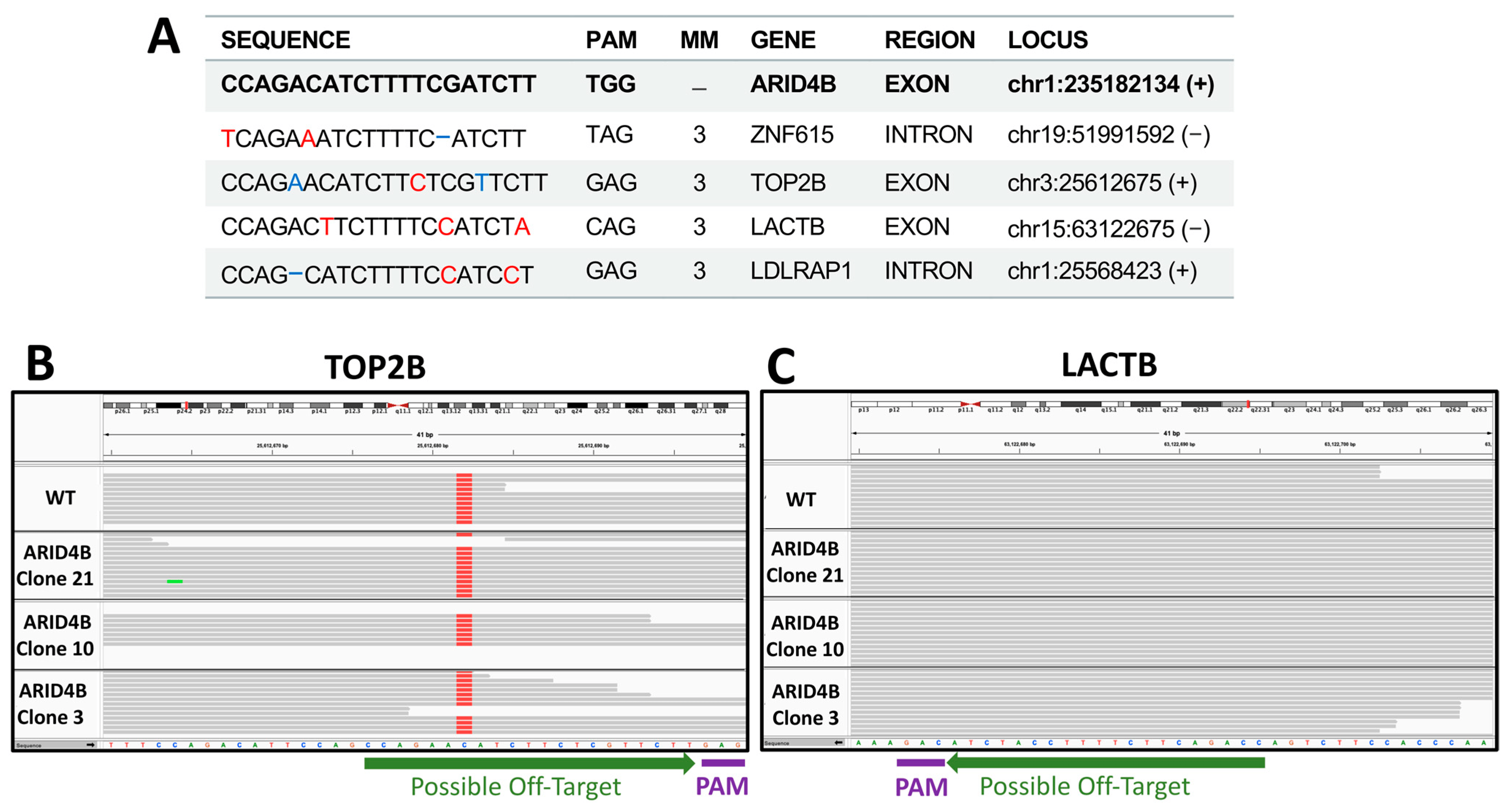
Figure 4. Off-targets validation. (A) Top possible off-target sequences. Red nucleotides mark mismatches with respect to the ARID4B sequence. Blue positions highlight insertions or deletions. Possible off-targets in exons are shown in B and C. (B) Exome sequencing of TOP2A possible off-target region in clones and wild-type cell lines. (C) Exome sequencing of LACTB possible off-target region in clones and wild-type cell line. No differences are observed relative to wild type (WT).
3.4. MCF-7 Clones Carrying Induced Mutations in ARID4B Show Reduced Cell Proliferation and Viability
To determine whether the induced mutation editions in ARID4B alter the proliferation and viability of BC cells, we measured the cell growth of clones ARID4B-3, ARID4B-10, and ARID4B-21 at different time points through the two following experiments. First, we used the Alamar Blue colorimetric test. Results show that our ARID4B clones are associated with reduced viability in MCF-7 cells at 48 h (p < 0.05, Figure 5A). Second, using the Cyquant Proliferation Kit, in which a reduction in cell proliferation was found from 48 to 96 h (p < 0.05) in the 3 clones compared with the NC (Figure 5B). Taking the results of both experiments, it was found that the ARID4B gene affects cell growth in MCF-7 cells.
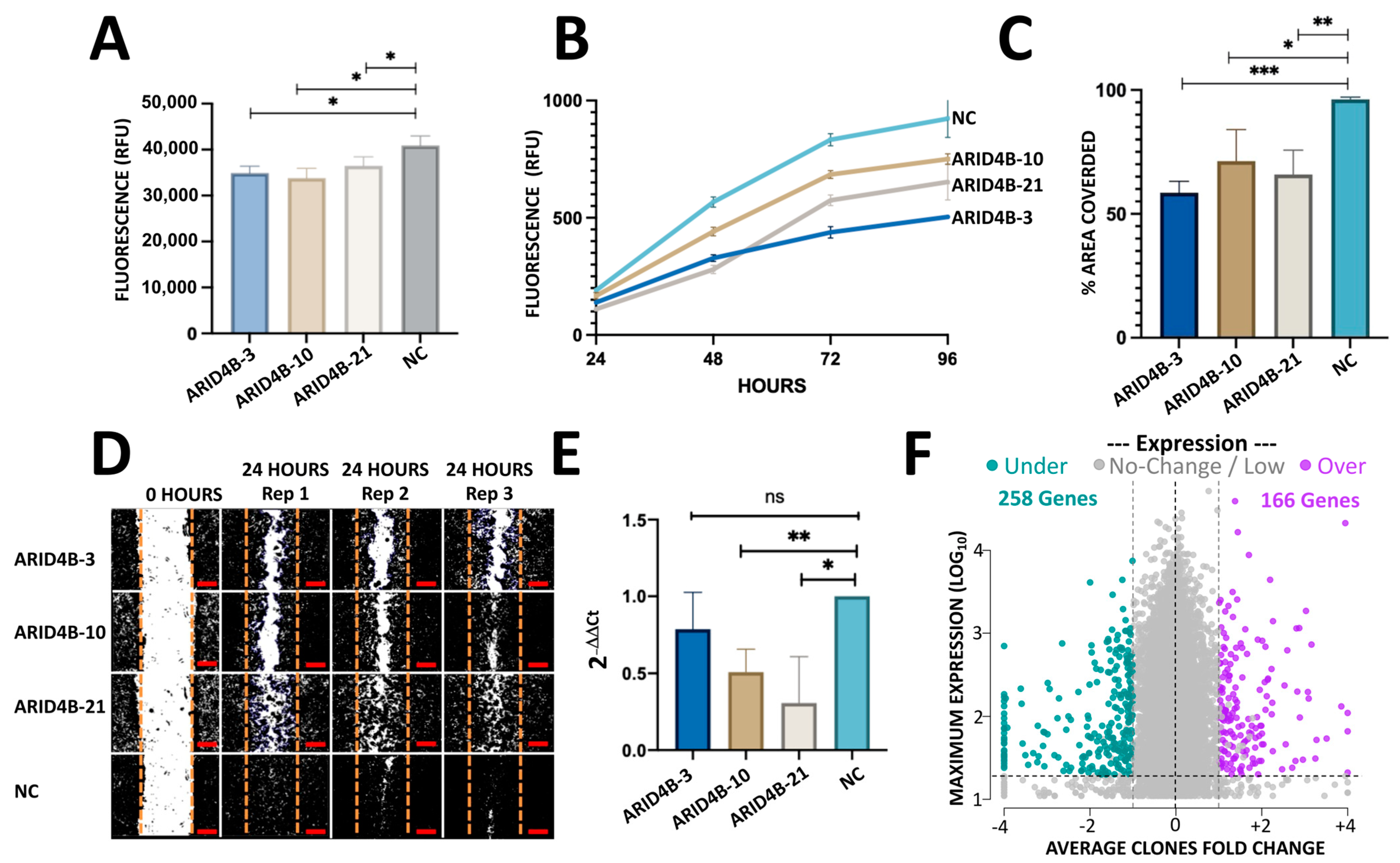
Figure 5. In vitro characterization of MCF-7 cells with mutations in ARID4B. (A) Viability assay with Alamar Blue shows a reduction in the three clones of ARID4B compared with NC. Results are shown as mean ± SEM (n = 3). (B) Mutations in ARID4B reduce proliferation using CyQuant Proliferation Kit. Results are shown as mean ± SEM (n = 3). (C,D) Reduction in cell migration in the three clones with mutations in ARID4B in a wound healing assay. Scale bars represent 100 µm. (E) qPCR analysis shows a reduction in expression in MCF-7 cells with mutations in ARID4B. (F) RNA-seq results gave a total of 424 genes up and down-regulated in cells with mutations compared with NC. The horizontal axis shows the average fold change from the 3 clones relative to NC. The vertical axis shows the maximum expression observed from the 3 clones and NC. In all cases * indicates p < 0.05, ** indicates p < 0.005 and *** indicates p < 0.0005.
3.5. Knockout of ARID4B Affects Migration Capacity
To evaluate whether ARID4B plays an essential role in cell migration, we assessed the wound-healing capacity of clones ARID4B-3, ARID4B-10, and ARID4B-21. Specifically, we imaged the area of the culture well that corresponded to the induced wound (which had been scratched with a pipet tip 24 h before imaging); then, we analyzed images using Image J and measured the percentage of area covered by each clone, relative to NC after 1 day of incubation. All three clones carrying mutations in ARID4B showed a decreased ability to migrate and cause “wound” closure, compared to NC (p < 0.05, Figure 5C,D). These results suggest that the presence of loss-of-function mutations in ARID4B is associated with lower migration rates in MCF-7 BC cells, potentially attenuating the metastatic capacity of BC cells.
3.6. Induction of Loss-of-Function Mutations Leads to Transcriptional Alterations of ARID4B
Using qPCR, a reduction in ARID4B expression levels was found in cells with loss-of-function mutations compared to wild-type cells. Specifically, clone ARID4B-21 was the clone that had a lower expression compared to NC, followed by ARID4B-3 and ARID4B-10 (Figure 5E). The order of expression level is very similar to that observed in RNA-seq data (31.7, 80.5, 121.4 transcripts per million in ARID4B-21, -10, and -3 clones, respectively).
3.7. Heterozygous Loss-of-Function Mutations in ARID4B Alter the Expression of Several Genes
We analyzed the transcriptome effect of the three clones by RNA-seq experiments. After read-count normalization, averaging across clones, and filtering by minimal counts and +/−1 fold-change in all clones relative to NC, a total of 166 genes were found to be overexpressed and 258 downregulated (Figure 5F). The top 20 genes are shown in while the whole results are available in (raw and processed data, PCA, and Heatmap in ). To further investigate the biological functions of these 424 deregulated genes, we performed 2 gene enrichment analyses using EnrichR and configuring different databases such as KEGG, Reactome, Bioplanet, and WikiPathways. The lists of up- and down-regulated genes were analyzed separately. To simplify the associations from different databases, we merged similar concepts and gene content in a “simplified concept”, as shown in (see for raw results). Some of the pathways that are enriched with the up-regulated genes are associated with cancer, such as epithelial to mesenchymal transition in colorectal cancer (FDR = 6%), PI3K-AKT signaling pathway (FDR = 2%) and focal adhesion-PI3K-AKT-mTOR (FDR = 8%). Other general significant pathways are amino acid biosynthesis (FDR < 1%), ECM receptor interaction (FDR = 0.9%), collagen metabolism, cytochrome P450, NCAM, and Platelet Signaling, among others .
On the other hand, down-regulated genes are associated with cholesterol biosynthesis (FDR ≤ 1%), metabolism of steroids (FDR ≤ 1%), TGF-β regulation of extracellular matrix (FDR ≥ 0.01%), and target of rapamycin (TOR) (FDR = 3%) . These results suggest that ARID4B could exert an important role in regulating genes associated with cancer progression.
4. Discussion
The ARID gene family is involved in regulating gene transcription and modifying chromatin structure [37]. Some members have been associated with cancer since these genes are frequently mutated in tumors and are related to cancer pathways [38]. In this work, we analyzed ARID4B specifically, which is involved in different functions, such as regulating male fertility [39] and spermatogenesis [40]. However, there are several studies related to the role of ARID4B and its participation in the development of different types of cancer. For example, in human glioma, it is highly expressed in cancer cell lines compared to normal brain tissue, and the higher expression correlates with WHO grade in patients [7]. On the other hand, ARID4B has been associated with tumor promoter functions regulated by microRNAs in prostate cancer [41], and others associate it as a tumor suppressor in leukemias [18]. In breast cancer, studies have been performed on mouse cells in which an ectopic expression of the ARID4B gene has been introduced, and an increase in proliferative and migratory capacities has been found. [19]. Furthermore, with the help of knockdown methodologies, a reduction in metastasis has also been demonstrated, which indicates that ARID4B would also have an important role in this type of cancer [19]. We did not find differences in the expression of ARID4B in breast cancer samples compared to normal tissues (Figure 1A), in contrast to what has already been observed in other types of cancer, such as hepatocellular [16] and human gliomas [42]. However, as previously documented in databases, worse survival was found in patients with a high expression (Figure 1B) [43]. This could indicate that the expression of ARID4B, specifically in BC, is associated with a more aggressive phenotype.
On the other hand, we also found that point mutations in ARID4B are associated with reduced survival in BC patients (Figure 1C). This evidence suggests that these mutations could be generating an over-activation. We recently found an important hotspot in ARID4B position 939 involving four cancer types (uterine, stomach, colorectal, and thymus). The hotspot is characterized by frame shifts that could be related to cancer progression also in BC. Thus, we decided to edit this region to find the effect of loss-of-function mutations generating heterozygous knockouts in MCF-7 cells using the CRISPR/Cas9 system. Our results clearly show that ARID4B frame-shift mutations around amino acid 939 reduce aggressiveness. Thus, the mutations observed decreasing survival in BC, which should correspond to gain of functions or loss-of-function before hotspot 939.
Since the function of ARID4B seems important in several cancer types, this study aimed to analyze the effect of loss-of-function in human breast cancer cells and to evaluate the pathways deregulated by the impact of the loss-of-function of the ARID4B gene. Our in vitro experiments revealed decreased viability, proliferation, and migration in clones with loss-of-function mutations compared to the negative control. Similar results have been found in other types of cancer, such as prostate and glioma mentioned above. For this reason, this study supports the hypothesis that the ARID4B gene is associated with promoting cancer-associated characteristics. Therefore, it would be interesting to observe other mutations that could explain the clinical observation.
Of the three clones obtained, ARID4B-3 and ARID4B-21 have a deletion, which would produce a truncated 939 amino acidic protein, while clone ARID4B-10, on the other hand, has an insertion that could produce a 936 amino acidic protein. Even though there is only a putative difference of three amino acids, the phenotypic assays revealed some differences between both types of mutations. The ARID4B-10 clone revealed a more significant negative effect on proliferation and migration. In contrast, the other two clones had similar results in both experiments. We noted an agreement in migration and proliferation across clones, suggesting that migration could be influenced by proliferation. In all cases, the mutations produced were heterozygous (partial knockout), which seems more similar to the genomic context in breast cancer patients in comparison to previous experiments performed in ARID4B that used RNA silencing or knockouts deleting both alleles and cell lines other than breast-derived [17,19,42]. In this way, the results of the in vitro characterization could explain the behavior of cells in BC development in a more specific manner.
In a systematic review, we have reported that the MCF-7 cell line is the most widely used model in breast cancer, studying mutations by CRISPR/Cas9 [44]. MCF-7 represents Luminal A type of breast cancer [45]. However, there are other cell lines that could display different behavior in breast or other cancer types. Thus, our analysis shows a fraction of the effects that ARID4B mutations could develop.
Employing RNA-Seq, the genes deregulated on edited cells were analyzed. We found the AKT-PI3K signaling through diverse collagens, RELN, LPAR5, GNG7, and ITGA5. AKT-PI3K is a critical pathway related to EGFR that regulates proliferation and cell survival [46]. The AKT-PI3K pathway has been previously linked with ARID4B in prostate cancer [17] and glioma [42] including downregulation of PI3KCA and PI3KR2. In contrast, we observed downregulation in PI3KC2A, PI3KC2B, PI3KC2G, and PI3KR3 (fold changes −1.6, −1.6, −5.4, and −1.2, respectively). Thus, our lesser cell proliferation agrees with other models but through different genes of the pathway, confirming and expanding our knowledge of the functional effects of ARID4B loss of function.
We also found pathways associated with the epithelial-to-mesenchymal transition (EMT), an important program that promotes the invasiveness of cells and leads to metastasis [47], and the EMT receptor interaction pathway, which has a critical role in cancer cells movement, adhesion, and hyperplasia [48]. Because other genes in the ARID family have been related to EMT pathways, for example, ARID1A [49] and JARID1B [50], our results suggest that ARID4B also has a role in modulating EMT. Its involvement could explain the differences in the migratory capacity found in the wound healing assay in this study and the metastasis observed in in vivo experiments published before [19].
Among the down-regulated pathways, two of the most enriched were cholesterol metabolism and the mevalonate pathway. Although no previous studies link ARID4B and these pathways, another ARID family member, ARID1A, showed an association by knockout experiments [51]. Comparatively, we noted similar genes downregulated such as IDI1, SQLE, HMGCS1, CYP51A1, DHCR24, MVD, HMGCR, and LSS (fold changes −1.7, −1.9, −2.3, −1.3, −1.2, −2.1, −1.4 and −2.0, respectively). This result suggests that both or more members of the ARID family are involved in regulating these pathways.
Another important altered pathway was the TGF-β regulation, which has been previously implicated with aggressive characteristics of breast cancer modulated by long non-coding RNAs [52,53,54]. In fact, blocking TGF-β can be very useful in developing therapies to inhibit tumor growth and metastasis [55]. ARID1A knockdown in MMNK-1 cells results in deregulated TGF-β members and increased proliferation and colony formation [56]. Thus, the expression or function of ARID1A and ARID4B genes seem to have opposite associations in cancer development. However, it is striking that the deregulation of the TGF-β signaling pathway also produces opposite results. This is consistent with other studies where TGF-β signaling in early cancer stages has a tumor suppressor effect, while in late stages, it has a promoter effect in proliferation and metastasis [55]. Here, using MCF-7 cells, the loss of function of ARID4B seems to deregulate genes of the TGF-β signaling pathway in both up- and down-regulated genes, which, by our functional assays, seems consistent with a reduction of aggressiveness. A study to evaluate the loss of function of ARID1A and ARID4B in both normal and cancer cells is necessary to verify specific effects on the TGF-β pathway, such as adding exogenous TGF-β in a rescue study.
A potential limitation of this study was the lack of funding to perform experiments analyzing Arid4b peptide expression. Although we showed enough evidence of mutated alleles being transcribed and that there is a reduction of gene expression, protein experiments could provide information on the relative amounts of allele-specific peptides and perhaps support the observed differences in clones 3 and 21. In addition, since results show an intriguing agreement in proliferation and migration, with the migration assay used, we can not rule out the influence of proliferation in the migration assays. Thus, more specific assays need to be performed to bring light to the specific role of ARID4B in these two processes.
5. Conclusions
The present study demonstrated that loss-of-function mutations close to a frequently mutated position of the ARID4B gene diminish its expression and alter the transcriptome program, reducing some aggressive characteristics of breast cancer, such as proliferation, viability, and migration. Therefore, it is likely that somatic mutations observed in patients showing worse prognosis and different frameshifts around amino acid 939 may confer more aggressive features, which should be demonstrated in further experiments. In addition, the RNA-Seq analysis performed in ARID4B clones revealed 410 dysregulated genes affecting different pathways, some associated with cancer and metastasis.
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding Driver Mutations in Cancer: Elucidating the Role of Background Mutational Processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hueng, D.Y.; Nieh, S.; Gao, H.W. ARID4B Is a Good Biomarker to Predict Tumour Behaviour and Decide WHO Grades in Gliomas and Meningiomas. J. Clin. Pathol. 2017, 70, 162–167. [Google Scholar] [CrossRef]
- Wilsker, D.; Probst, L.; Wain, H.M.; Maltais, L.; Tucker, P.W.; Moran, E. Nomenclature of the ARID Family of DNA-Binding Proteins. Genomics 2005, 86, 242–251. [Google Scholar] [CrossRef]
- Cao, J.-N.; Gao, T.-W.; Stanbridge, E.J.; Irie, R. RBP1L1, a Retinoblastoma-Binding Protein-Related Gene Encoding an Antigenic Epitope Abundantly Expressed in Human Carcinomas and Normal Testis. JNCI J. Natl. Cancer Inst. 2001, 93, 1159–1165. [Google Scholar] [CrossRef]
- Wu, M.Y.; Tsai, T.F.; Beaudet, A.L. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b Alters Epigenetic Modifications and Suppresses an Imprinting Defect in the PWS/AS Domain. Genes Dev. 2006, 20, 2859–2870. [Google Scholar] [CrossRef]
- Treviño, V.; Tamez-Pena, J. VALORATE: Fast and Accurate Log-Rank Test in Balanced and Unbalanced Comparisons of Survival Curves and Cancer Genomics. Bioinformatics 2017, 33, 1900–1901. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refines Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Mamo, A.; Cavallone, L.; Tuzmen, S.; Chabot, C.; Ferrario, C.; Hassan, S.; Edgren, H.; Kallioniemi, O.; Aleynikova, O.; Przybytkowski, E.; et al. An Integrated Genomic Approach Identifies ARID1A as a Candidate Tumor-Suppressor Gene in Breast Cancer. Oncogene 2012, 31, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Guo, T.; Chua, P.J.; Tang, L.; Thike, A.A.; Tan, P.H.; Bay, B.H.; Baeg, G.H. Clinicopathological Significance of ARID1B in Breast Invasive Ductal Carcinoma. Histopathology 2015, 67, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, Z.; Chen, F.; Liao, C.; Wang, Q.; Huang, X. Overexpression of ARID4B Predicts Poor Survival in Patients with Hepatocellular Carcinoma. Hum. Pathol. 2018, 73, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Young, I.C.; Chen, Y.F.; Chuang, S.T.; Toubaji, A.; Wu, M.Y. Identification of the PTEN-ARID4B-PI3K Pathway Reveals the Dependency on ARID4B by PTEN-Deficient Prostate Cancer. Nat. Commun. 2019, 10, 4332. [Google Scholar] [CrossRef]
- Wu, M.Y.; Eldin, K.W.; Beaudet, A.L. Identification of Chromatin Remodeling Genes Arid4a and Arid4b as Leukemia Suppressor Genes. J. Natl. Cancer Inst. 2008, 100, 1247–1259. [Google Scholar] [CrossRef]
- Winter, S.F.; Lukes, L.; Walker, R.C.; Welch, D.R.; Hunter, K.W. Allelic Variation and Differential Expression of the MSIN3A Histone Deacetylase Complex Gene Arid4b Promote Mammary Tumor Growth and Metastasis. PLoS Genet. 2012, 8, e1002735. [Google Scholar] [CrossRef]
- Goldberger, N.; Walker, R.C.; Kim, C.H.; Winter, S.; Hunter, K.W. Inherited Variation in MiR-290 Expression Suppresses Breast Cancer Progression by Targeting the Metastasis Susceptibility Gene Arid4b. Cancer Res. 2013, 73, 2671–2681. [Google Scholar] [CrossRef]
- Trevino, V. HotSpotAnnotations—A Database for Hotspot Mutations and Annotations in Cancer. Database 2020, 2020, baaa025. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Salinas, F.; Rojo, R.; Martinez-Amador, C.; Herrera-Gamboa, J.; Trevino, V. Transcriptomic and Cellular Analyses of CRISPR/Cas9-Mediated Edition of FASN Show Inhibition of Aggressive Characteristics in Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2020, 529, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Rausch, T.; Fritz, M.H.Y.; Untergasser, A.; Benes, V. Tracy: Basecalling, Alignment, Assembly and Deconvolution of Sanger Chromatogram Trace Files. BMC Genom. 2020, 21, 230. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A Platform for Interactive Large-Scale Genome Analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In Vitro Scratch Assay: A Convenient and Inexpensive Method for Analysis of Cell Migration in Vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Boekel, J.; Chilton, J.M.; Cooke, I.R.; Horvatovich, P.L.; Jagtap, P.D.; Käll, L.; Lehtiö, J.; Lukasse, P.; Moerland, P.D.; Griffin, T.J. Multi-Omic Data Analysis Using Galaxy. Nat. Biotechnol. 2015, 33, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Zhu, Y.; Yan, C.; Wang, X.; Xu, Z.; Lv, J.; Xu, X.; Yu, W.; Zhou, M.; Yue, L. Pan-Cancer Analysis of ARID Family Members as Novel Biomarkers for Immune Checkpoint Inhibitor Therapy. Cancer Biol. Ther. 2022, 23, 104–111. [Google Scholar] [CrossRef]
- Lin, C.; Song, W.; Bi, X.; Zhao, J.; Huang, Z.; Li, Z.; Zhou, J.; Cai, J.; Zhao, H. Recent Advances in the ARID Family: Focusing on Roles in Human Cancer. Onco. Targets. Ther. 2014, 7, 315–324. [Google Scholar] [CrossRef]
- Wu, R.C.; Jiang, M.; Beaudet, A.L.; Wu, M.Y. ARID4A and ARID4B Regulate Male Fertility, a Functional Link to the AR and RB Pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 4616–4621. [Google Scholar] [CrossRef]
- Wu, R.C.; Zeng, Y.; Pan, I.W.; Wu, M.Y. Androgen Receptor Coactivator ARID4B Is Required for the Function of Sertoli Cells in Spermatogenesis. Mol. Endocrinol. 2015, 29, 1334–1346. [Google Scholar] [CrossRef]
- Liang, Y.; Han, Z.; Lu, J.; Liu, Z.; Zhuo, Y.; Zhu, X.; Chen, J.; Ye, J.; Liang, Y.; He, H.; et al. Downregulation of ARID4A and ARID4B Promote Tumor Progression and Directly Regulated by MicroRNA-30d in Patient with Prostate Cancer. J. Cell. Biochem. 2018, 119, 7245–7255. [Google Scholar] [CrossRef]
- Luo, S.M.; Tsai, W.C.; Tsai, C.K.; Chen, Y.; Hueng, D.Y. Arid4b Knockdown Suppresses Pi3k/Akt Signaling and Induces Apoptosis in Human Glioma Cells. Onco Targets Ther. 2021, 14, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, S.; You, Z.; Li, G.; Xu, S.; Li, X.; Zhang, X.; Lei, B.; Pang, D. Expression and Prognostic Values of ARID Family Members in Breast Cancer. Aging 2021, 13, 5621–5637. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Salinas, F.; Martinez-Amador, C.; Trevino, V. Characterizing Genes Associated with Cancer Using the CRISPR/Cas9 System: A Systematic Review of Genes and Methodological Approaches. Gene 2022, 833, 146595. [Google Scholar] [CrossRef]
- Moon, H.; Ospina-Muñoz, N.; Noe-Kim, V.; Yang, Y.; Elzey, B.D.; Konieczny, S.F.; Han, B. Subtype-Specific Characterization of Breast Cancer Invasion Using a Microfluidic Tumor Platform. PLoS ONE 2020, 15, e0234012. [Google Scholar] [CrossRef]
- Safdari, Y.; Khalili, M.; Ebrahimzadeh, M.A.; Yazdani, Y.; Farajnia, S. Natural Inhibitors of PI3K/AKT Signaling in Breast Cancer: Emphasis on Newly-Discovered Molecular Mechanisms of Action. Pharmacol. Res. 2015, 93, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.W. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J. Clin. Med. 2019, 8, 646. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, L.; Shi, L.; Yun, F.; Liu, X.; Chen, Y.; Chen, C.; Ren, Y.; Jia, Y. Transcriptome Profiling Revealed Multiple Genes and ECM-Receptor Interaction Pathways That May Be Associated with Breast Cancer. Cell. Mol. Biol. Lett. 2019, 24, 38. [Google Scholar] [CrossRef]
- Tomihara, H.; Carbone, F.; Perelli, L.; Huang, J.K.; Soeung, M.; Rose, J.L.; Robinson, F.S.; Lissanu Deribe, Y.; Feng, N.; Takeda, M.; et al. Loss of ARID1A Promotes Epithelial–Mesenchymal Transition and Sensitizes Pancreatic Tumors to Proteotoxic Stress. Cancer Res. 2021, 81, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Stadler, S.C. Role of Epigenetic Mechanisms in Epithelial-to-Mesenchymal Transition of Breast Cancer Cells. Transl. Res. 2015, 165, 126–142. [Google Scholar] [CrossRef]
- Goldman, A.R.; Bitler, B.G.; Schug, Z.; Conejo-Garcia, J.R.; Zhang, R.; Speicher, D.W. The Primary Effect on the Proteome of ARID1A-Mutated Ovarian Clear Cell Carcinoma Is Downregulation of the Mevalonate Pathway at the Post-Transcriptional Level. Mol. Cell. Proteom. 2016, 15, 3348–3360. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, S.H.A.; Cheng, H.; Li, J.; Feng, R. The Effect of TGF-β-Induced Epithelial-Mesenchymal Transition on the Expression of Intracellular Calcium-Handling Proteins in T47D and MCF-7 Human Breast Cancer Cells. Arch. Biochem. Biophys. 2015, 583, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Mehterov, N.; Ling, H.; Stanta, G.; Braicu, C.; Berindan-Neagoe, I. The “Good-Cop Bad-Cop” TGF-β Role in Breast Cancer Modulated by Non-Coding RNAs. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-J.; Li, Y.; Wu, Y.-Z.; Wang, Y.; Nian, W.-Q.; Wang, L.-L.; Li, L.-C.; Luo, H.-L.; Wang, D.-L. Long Non-Coding RNA CCAT2 Promotes the Breast Cancer Growth and Metastasis by Regulating TGF-β Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 706–714. [Google Scholar] [PubMed]
- Syed, V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Friedland, S.C.; Alexander, W.; Myers, J.A.; Wang, W.; O’Dell, M.R.; Getman, M.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; et al. Arid1a Mutation Suppresses TGF-β Signaling and Induces Cholangiocarcinoma. Cell Rep. 2022, 40, 111253. [Google Scholar] [CrossRef]
