


1. Introduction
Newborns, particularly preterm neonates, who are admitted to a neonatal intensive care unit (NICU) are highly susceptible to temporary or permanent colonisation of their skin and/or mucous membranes with multi-drug-resistant Gram-negative bacteria (MRGN). This is primarily due to the frequent administration of antibiotics and the extended duration of hospitalisation [1]. Importantly, premature infants face a higher likelihood of developing MRGN infections because of their immature immune state, especially when confronted with intestinal complications (e.g., necrotising enterocolitis) or healthcare-associated infections (HAIs, e.g., central venous catheters, parenteral nutrition, invasive ventilation and drains). Infectious material includes various substances such as respiratory secretions (including aerogenic transmission), faeces, urine and wound secretions, among others [2]. Thus, it is important to avoid the contamination of surfaces—especially of wet areas within the immediate vicinity of the patient’s environment (e.g., incubators, washbasins, syphons, taps, water baths, storage tanks of humidifiers, etc.). Surface contamination enables transmission via contaminated hands (parents/medical and nursing staff), material or food.
In Germany, MRGN are classified according to the guidelines from the Commission for Hospital Hygiene and Infection Prevention (KRINKO) of the Robert Koch Institute (RKI, Berlin, Germany): In adults, multi-drug resistance (MDR) of Gram-negative bacilli is defined as phenotypic resistance to ≥3 of 4 antibiotic classes ([1] piperacillin, [2] third generation cephalosporins, [3] flurochinolones and [4] carbapenems) or the detection of carbapenemases. However, in neonates, a separate class, so-called 2MRGN-NeoPaed, is defined, which exhibits phenotypic resistance to two out of the four antibiotic classes [3], as the use of fluoroquinolones for empirical therapy is not suitable in neonatology and paediatrics.
In addition to aiding decision-making in antibiotic stewardship (ABS), the early detection of MRGN status plays a crucial role in implementing patient isolation and cohorting strategies in NICUs, hence the German KRINKO-published recommendations on patient-related colonisation screening (CoS) for MRGN bacilli in NICU neonates, as of 2007 [4]. These involve obligatory nasopharyngeal (or throat) and rectal swabs to facilitate a calculated antibiotic therapy at an early stage in very low-birth-weight infants of all NICUs in Germany.
Most neonates are asymptomatic and not infected upon the first MRGN detection (colonised state). Noteworthy, the infection rate of previously colonised neonates depends not only on the host-specific risk factors associated with the premature state [5,6,7] but also on the bacterial species [8]. Typical Gram-negative infectious agents associated with late-onset sepsis in neonates are Enterobacter cloacae complex (ECC), Klebsiella pneumoniae and Serratia marcescens [5,8,9,10,11,12]. It is of note that the role of MRGN colonisation as a potential driver of the infection of preterm neonates at later stages is currently being controversially discussed [13,14,15].
Due to the close relationships within the ECC, antibiotic susceptibility patterns (ASTs) and routine matrix-assisted laser desorption–ionisation time of flight mass spectrometry (MALDI–TOF MS) can be misleading in indicating phylogenetic relatedness within the ECC in outbreak scenarios [16], as shown by previous studies [17,18,19,20,21,22]. Thus, high-resolution strain typing methods such as pulsed field gel electrophoresis (PFGE) or whole-genome sequencing (WGS) are required [19,23,24,25]. WGS offers superior resolution compared to the previous gold standard method of PFGE, allowing for the identification of outbreaks at subclonal levels. Furthermore, the use of next-generation sequencing (NGS) data and tools for phylogenetic analyses such as core genome multilocus sequence typing (cgMLST) or the detection of whole-genome single-nucleotide polymorphisms (wgSNPs) offers the opportunity to improve outbreak investigations by providing information on the microbial community composition of the hospital environment [26,27,28]. In our study, we describe the usage of bacterial WGS in combination with wgSNP analysis as a valuable tool for the management of an ECC outbreak on a German tertiary NICU.
2. Materials and Methods
2.1. Study Population
The Department of Paediatrics and Adolescent Medicine of the Friedrich-Alexander-University Erlangen-Nürnberg, Germany, includes a 14-bed level-3 neonatal intensive care unit (NICU) with an associated standard neonatal care unit of 16 beds. A level-3 NICU is a specialised healthcare facility that provides the highest level of specialised care for critically ill newborns, including those with complex medical conditions and those requiring advanced interventions. In 2020, the NICU had 799 admissions from both in-born and referred patients. Along with its associated children’s heart centre and pediatric intensive care unit, it functions as a reference centre for other hospitals in the Franconia province of Bavaria. Bacterial screening (upon submission and then weekly) of all neonates in the NICU has been an implemented routine (with nasal and rectal swabs, as well as tracheal aspirates for intubated/tracheotomised neonates) since 2012.
The outbreak commenced in April 2020 and was resolved by June 2020. During that period, 10 neonates showed colonisation with ECC. Retained frozen samples of 10 of these strains were further analysed using NGS (see below).
Preventive measures during the outbreak aimed to reduce nosocomial transmission and included contact precautions such as isolating MRGN neonates in single rooms whenever possible, using disposable gowns and nitrile gloves for the handling of all neonates and supervising the use of alcoholic hand disinfection (Desderman, Schülke & Mayr GmbH, Norderstedt, Germany). The dedicated assignment of specific nursing staff to the MRGN patient cohort was strictly implemented. Staff compliance with hygiene standards and environmental cleaning was also monitored. Despite conducting systematic line list analyses of medical and nursing staff and intensive microbiological examination of the extended patient environment, the results were inconclusive. However, a new standardised operating procedure was implemented to improve the handling of MRGN-colonised neonates by medical professionals and parents (see ).
2.2. Strain Isolation and Microbiological Characterisation
All strains were isolated from the rectal swabs obtained for screening purposes as described previously [13]. Strain identification was done using MALDI–TOF MS (Bruker Daltonik GmbH, Bremen, Germany) and AST was performed using a VITEK 2 system (bioMérieux, Nürtingen, Germany). The latest EUCAST breakpoints (v10.0, 2020) were utilised for interpretation whenever possible.
2.3. Whole Genome Sequencing and Assembly
The genomic DNA for bacterial WGS was isolated from 2 mL of overnight culture in lysogeny broth (LB) using the GenElute Bacterial Genomic DNA kit (Sigma-Aldrich, Schnelldorf, Germany) according to the manufacturers’ instructions. The DNA was adjusted to 0.2 ng/µL in 10 mM of Tris-HCl pH 8.5 and 2.5 µL and subjected to tagmentation using the Nextera XT kit (Illumina, Berlin, Germany). The libraries were dual-indexed using PCR and the Nextera index set A (Illumina) and the mean fragment size was determined on a TapeStation 4200 instrument (Agilent, Waldbronn, Germany). The libraries were adjusted to 7 pM and subjected to 2 × 250 bp paired-end sequencing on a MiSeq instrument (Illumina, Berlin, Germany). Sequence data were demultiplexed according to the index sequences with the “Generate Fastq” workflow in MiSeq Reporter (Illumina) and are available in the National Centre for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/ accessed on 12 October 2023) Sequence Read Archive (SRA) under BioProject PRJNA727521. The adapter sequences were removed, and the sequence data were filtered based on quality using BBDuk from BBMap v38.79 (Joint Genome Institute, https://sourceforge.net/projects/bbmap/ accessed on 12 October 2023). SKESA v2.3.0 [29] was applied for de novo assembly from filtered short reads. Species assignment was carried out based on the average nucleotide identity with GenBank type strains [30]. The assignment of isolates to Enterobacter hormaechei subspecies [31] was completed using MASH v2.2.2 [32] with a sketch size of 10,000 and k = 21 from the following reference genomes: CP017186.1 (subsp. hoffmannii), CP010377.1 (subsp. hormaechei), CP017180.1 (subsp. oharae), CP017179.1 (subsp. steigerwaltii) and CP017183.1 (subsp. xiangfangensis).
2.4. Phylogenetic Analysis
A contig-based whole-genome SNP alignment was done using PhaME v1.0.2 [33] and using ECC strain AR_0060 (GenBank: CP026719.1) as a reference. An approximately maximum likelihood (ML) tree was calculated for the isolates with FastTree 2 [34]. The tree was further processed and visualised using the GGTREE package for R [35].
2.5. Detection of Antimicrobial Resistance Genes and Mobile Genetic Elements
Genes conferring antimicrobial resistance (AMR) were identified in the assembled genomes using ABRicate v1.0.1 (Seemann T, https://github.com/tseemann/abricate accessed on 12 October 2023) and the NCBI AMRFinderPlus database (5684 entries after filtering, accessed 7 May 2021) [36]. The results were summarised with the summary option built into ABRicate and the isolates were clustered using an unweighted pair group method with arithmetic mean (UPGMA) based on a Jaccard distance matrix calculated with the antimicrobial-resistant gene presence using a custom script in R v4.2.2 [37]. MOB-suite [38] was used to detect mobile genetic elements (MGEs) as well as to reconstruct and type the plasmids from the de novo-assembled genomes [38,39,40].
3. Results
3.1. Patient Characteristics
We analysed perianal skin swabs from 6 male and 4 female neonates. The median gestational age at birth was 32 + 4 weeks (minimum 24 + 0, maximum 39 + 5 weeks). The interquartile range (IQR) of the birth weight was 787.5 to 2378 g with a median of 1585 g. The median chronological age at the onset of first MRGN detection was 21.5 days (IQR: 13 to 54.5 days). The median length of NICU stay was 60.5 days (IQR: 28.0 to 82.5 days).
Our retrospective analysis of NeoKiss bacterial surveillance revealed a significant increase in the total prevalence of 2MRGN-NeoPaed bacteria (20 cases per 100 neonates) in our NICU during the MRGN outbreak of April to June 2020. This was above the national median of 3.60 (75%-quantile 8.21) for that year. Additionally, the incidence of nosocomial-acquired multidrug-resistant bacteria (18.18 cases per 100 neonates) was also significantly higher than the national median of 3.12 cases (75%-quantile 7.52), with an incidence density of 6.93 nosocomial cases per 1000 patient days (national median: 1.02, 75%-quantile 2.19). In 2020, the majority of 2MRGN-NeoPaed (90.91%) and 3MRGN (100%) cases were acquired nosocomially (55 cases, 1443 patient days). Although the infection frequency for the identified MRGN colonisers was generally low in our NICU [13], two de novo nosocomial infections with 2MRGN-NeoPaed were identified in 2020, but they were not related to the analysed outbreak.
3.2. Characterization of Bacterial Isolates
Phenotypic AST and strain identification were carried out as part of the microbiological routine diagnostics. All isolates were identified as ECC since the current routine MALDI–TOF MS approaches fail to reliably identify these bacteria at the species level [41]. All isolates were classified as 2MRGN-NeoPaed based on phenotypic resistance to penicillins (piperacillin) and cephalosporins (ceftriaxon). Overall, the isolates exhibited a very homogeneous resistance pattern. This was also true for the minimal inhibitory concentrations (MIC) of fosfomycin (Fos) and tetracycline (Tet), where both antibiotics differed by only one dilution step, except for strains VA41244 and VA42547. These strains showed a lower MIC of ≤16 mg/L for Fos and isolate VA42547 was more susceptible to Tet having a MIC of ≤1 mg/L .
For WGS, the genomes were constructed using de novo assembly from short reads resulting in a median of 39 contigs per isolate with a median of 111 for the mean read coverage . The contigs were included in a wgSNP alignment with ECC strain AR_0060 as reference. The resulting approximately ML tree exhibited a topology with three clearly separated clades: The first clade consisted of six clonal isolates (VA33829, VA33831, VA33836, VA34552, VA34560, VA35386), the second of a single isolate (VA42547) and the third clade of two clonal strains (VA33843, VA36175) and one related isolate (VA41244) (Figure 1). In the next step, we determined the Enterobacter species and subspecies of all isolates based on the assembled genomes. Species identification was undertaken with MASH [32] calculating hashes based on the MinHash sketches derived from small oligonucleotides (k-mers), which can be very efficiently used for comparative analyses. Here, a set of Enterobacter species and subspecies reference genomes was used for comparison. We found all isolates were Enterobacter hormaechei including three subspecies: subsp. steigerwaltii (n = 6, 60%), subsp. hoffmannii (n = 3, 30%) and subsp. oharae (n = 1, 10%) . As expected, subspecies assignment correlated perfectly with the three clades previously identified in the wgSNP-based approximately ML tree (Figure 1).
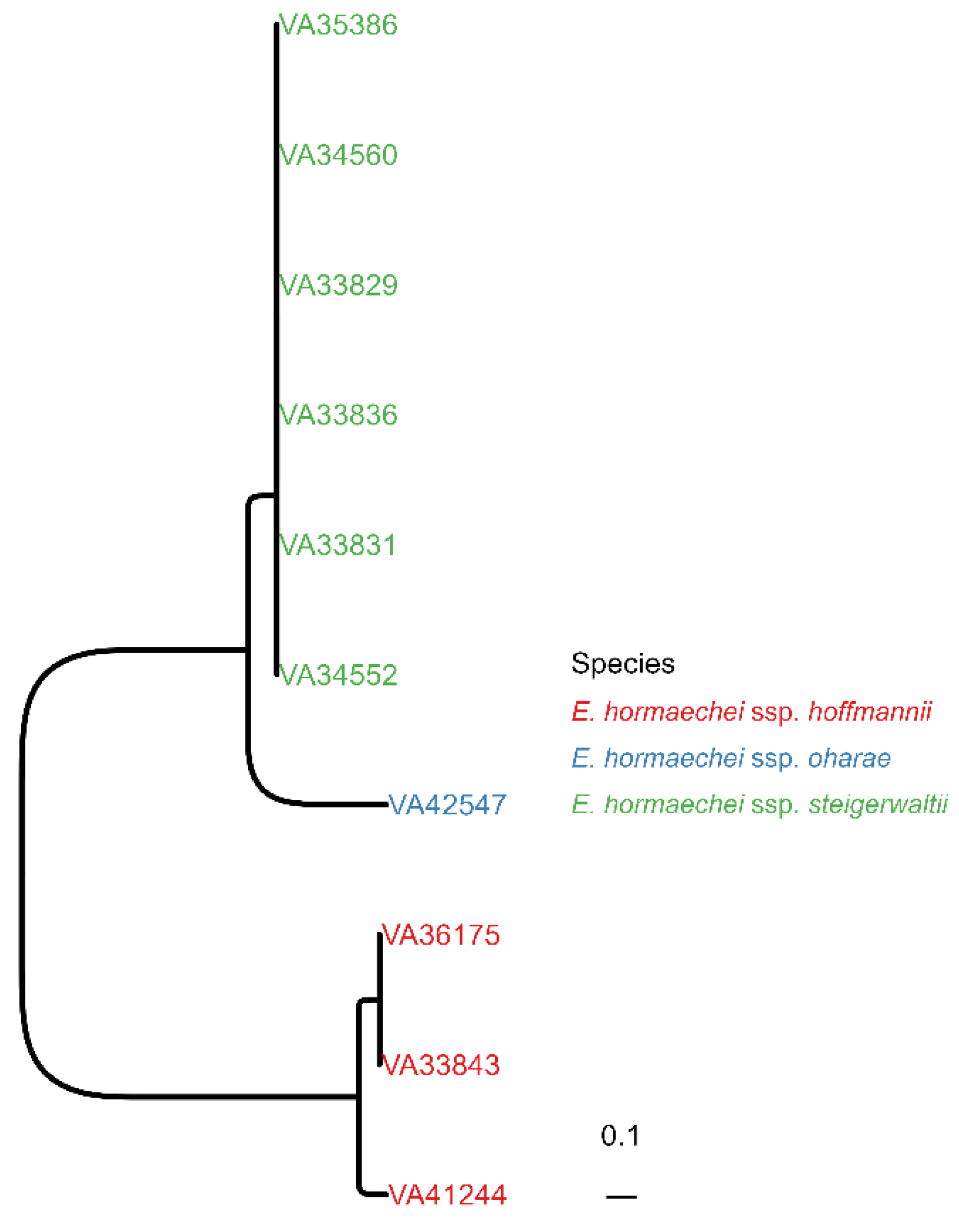
Figure 1. Phylogenetic relationship between E. hormaechei isolates. Approximately maximum likelihood (ML) tree from contig-based whole-genome single-nucleotide polymorphism (wgSNP) alignment is shown. The isolates were coloured according to their subspecies assignment.
Further, the contigs of the bacterial isolates were screened for AMR genes, MGEs and plasmids. BLAST was used to screen for the presence of the AMR genes listed in the NCBI AMRFinderPlus database. Within the genomes of the E. hormaechei isolates, we identified a total of 10 AMR genes that had significant homologs (Figure 2). All isolates carried the class C β-lactamases responsible for conferring resistance against the penicillin/β-lactamase inhibitor combinations ampicillin/sulbactam and piperacillin/tazobactam, as well as against second- (Cefuroxime) and third (Ceftazidime)-generation cephalosporins . Interestingly, the β-lactamase subtypes identified correlated with the subspecies. ACT-40 was found in all subsp. steigerwaltii, ACT-55 in the subsp. oharae while the two closely related subsp. hoffmannii (VA33843, VA36175) carried ACT-5 and the other subsp. hoffmannii (VA41244) ACT-67. Furthermore, genes for both subunits of the OqxAB multidrug resistance-nodulation-division (RND) efflux transporter were detected in all isolates. Here, for subsp. oharae and subsp. Steigerwaltii, the alleles oqxA9/oqxB9 were detected, whereas for subsp. hoffmannii strains, different combinations of oqxA10 with oqxB15 (VA33843, VA36175) or oqxB5 (VA41244) were found. Although two of the strains exhibited decreased resistance against fosfomycin (MIC ≤ 16 µg/mL, , the gene for Fos resistance-conferring glutathione transferase FosA was present in 9 out of 10 E. hormaechei isolates. Strain VA42547 (subsp. oharae) lacked the fosA gene, which was in accordance with its low fosfomycin MIC.
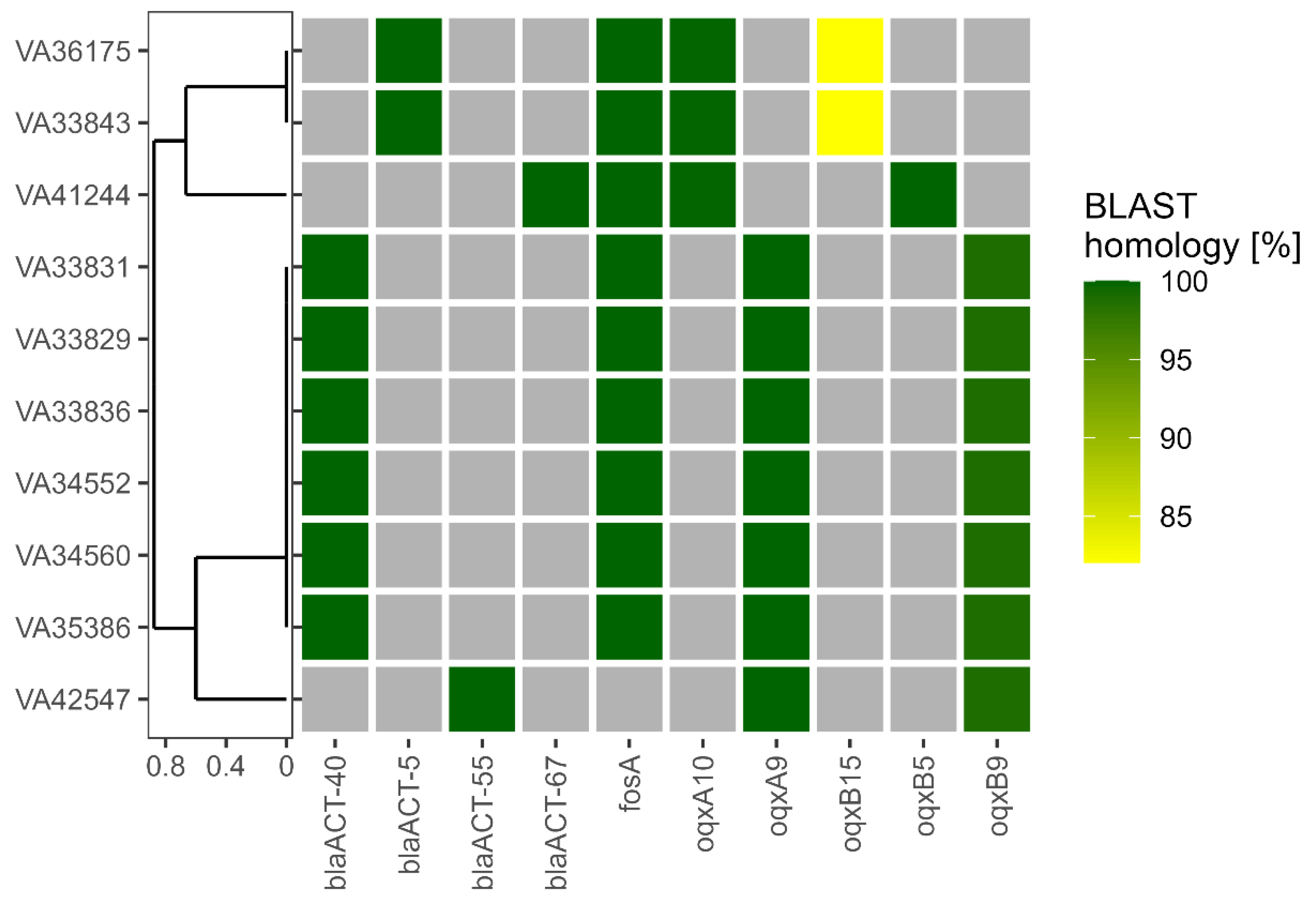
Figure 2. Detection of antimicrobial resistance (AMR) genes. AMR genes were identified in the assembled genomes of the isolates indicated in a Basic Local Alignment Search Tool (BLAST)-based approach. Coloured squares indicate the presence of the respective AMR gene with shading between yellow and green depending on BLAST homology. Gray squares show the absence of the respective AMR gene. The isolates were ordered according to a tree generated using unweighted pair group method with arithmetic mean (UPGMA) based on a Jaccard distance matrix derived from AMR gene presence/absence as shown here. Legend: blaACT gene—coding for a C-type beta-lactamase; fosA gene—coding for a glutathione-S-transferase that inactivates fosfomycin; oqxA gene—coding for a resistance-nodulation-cell division (RND) efflux pump conferring resistance to fluoroquinolone.
A significant concern lies in the dissemination of antibiotic resistances within hospital environments. Because mobile genetic elements (MGEs) such as plasmids, phages or transposons play a crucial role in the intercellular mobility of AMR genes [42], we identified MGEs and their spatial correlation with AMR genes in all isolates. An overview of the predicted plasmids and insertion (IS) elements is given in . All isolates except VA34560 harboured a derivative of a small (~2.5 kb) plasmid of the ColE1 replicon type, which is widely distributed amongst Enterobacter and Klebsiella. Furthermore, we identified in all strains except VA41244 and VA42547 a larger (~4.7 kb) plasmid having the mobBCD mobilization genes only. Strain VA33843 carried in addition a large ~109 kb plasmid of the IncF replicon type, harbouring two IS elements with highest similarity to E. hormaechei subsp. hormaechei ECR091 plasmid pENT-4bd (accession: CP008907). A total of six IS elements were identified in strain VA41244: while one was encoded on plasmid p4, two were found on the genome, and four different IS elements on the larger plasmid p6. It is noteworthy that, based on the contig data, the identified AMR genes were neither found on the predicted plasmids nor in the vicinity of the chromosomal IS elements (Figure 2).
4. Discussion
The utilisation of whole-genome sequencing (WGS) and subsequent bioinformatic analysis has been widely accepted as an effective approach to strain typing in clinical microbiology and outbreak investigations [19,43,44], particularly for members of the Enterobacteriaceae family [19,45,46]. Therefore, we employed bacterial WGS in a neonatal intensive care unit (NICU) outbreak to establish the phylogenetic connection among the ECC isolates. This information served as a foundation for enhancing our isolation strategies moving forward.
While bacterial screening via environmental examinations (surface areas, sinks, etc.) was inconclusive, we were able to identify three E. hormaechei subspecies, mainly ssp. steigerwaltii, but also ssp. hoffmannii and a single case of ssp. oharae, as bacterial colonisers in our neonates.
According to Sutton et al. [31], E. hormaechei is the most prevalent species among the clinical isolates of ECC. However, as mentioned earlier, distinguishing between different species within ECC using current routine MALDI–TOF MS systems is not feasible. This limitation could potentially be resolved by utilising enhanced databases [47].
Historically, E. hormaechei was suggested in 1989 as a new member of the Enterobacter genus [48]. During the last three decades, E. hormaechei has emerged as a relevant nosocomial pathogen in hospitalised adults [31], as well as an opportunistic pathogen in NICUs.
In 1997, Wenger et al. reported a case series involving six instances of E. hormaechei bloodstream infections and four cases of colonisation of preterm infants at the Hospital of the University of Pennsylvania, USA. The affected neonates had a low gestational age and birth weight. The transmission of E. hormaechei within the hospital setting may have been facilitated by environmental contamination, such as the contamination of neonatal incubator systems and doorknobs, as well as lapses in infection control practices among healthcare workers [49].
Da Silva et al. described six cases of neonates with E. hormaechei bloodstream infection in three NICUs in Rio de Janeiro, Brazil, in 2002 with complete recovery under antibiotic treatment. While the source remained unclear, parenteral nutrition was identified as the only common procedure [50]. In contrast, Dyabi et al. reported a series of three fatalities among five E. hormaechei bloodstream-infected neonates (mostly preterm) at the NICU of Mohamed VI University Hospital, Marrakesh, Morocco, in 2018. Most patients presented with respiratory distress as a common clinical sign [49].
Girlich et al. analysed a large incubator-related outbreak in the NICU of Bicêtre Hospital, Paris, France, with around 30% of neonates being physiologically colonised with ECC. Seven neonates had bacteraemia, and 6 of them were lost to fatal sepsis, systematically linked to (hypervirulent) E. bugandensis [51]. This study highlights the need for the better discrimination of Enterobacter species inside the ECC in this fragile population, as neonates colonised or infected with other ECC species had favourable outcomes in this study.
In line with these reports, E. hormaechei colonisation in our study affected mostly preterm neonates during the neonatal period (<28 d). Although phylogenetic analyses revealed the involvement of four different clones, E. hormaechei subsp. steigerwaltii was the dominant outbreak clone (n = 6). However, the environmental contribution remained inconclusive and general improvement of infection control techniques among health workers and parents (see ) resolved the issue. While the outbreak-driven increase in ECC screening isolates was evident in 2020, we observed a drop to normal levels in the following year (Figure 3). Therefore, this aspect seems to be an important mainstay of hospital hygiene regarding the sustainable containment of ECC in general and particularly of E. hormaechei. It has been shown that microbial communities on surfaces in patient rooms closely resemble the respective skin microbiome [26,52]. In NICUs, the predominant bacterial genera were Streptococcus and Staphylococcus [53]. Martineau et al. also pointed out the strong effect of surface disinfection on bacterial communities in the case of Serratia marcescens [19], as also described by others [28]. The fact that we did not identify these bacterial communities in our patient environment might imply sufficient surface hygiene, further supporting the human-to-human transmission of E. hormaechei during our outbreak despite inconclusive line list analyses.
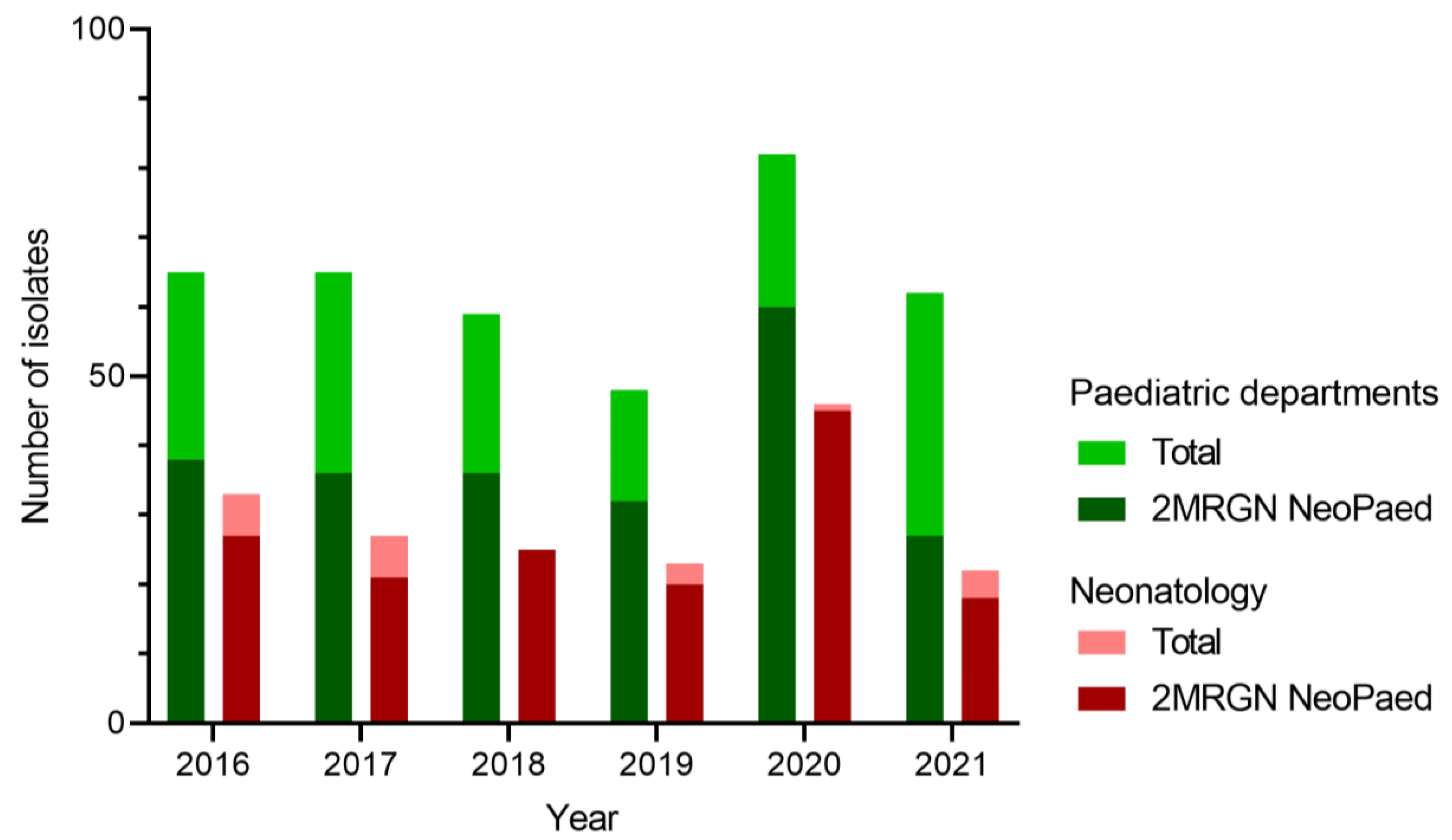
Figure 3. Numbers of E. cloacae complex (ECC) isolates in screening materials per year. In green, the number of annual ECC isolates from all paediatric departments of the University Hospital Erlangen are shown. In dark green the proportion of 2MRGN NeoPaed-classified ECC is depicted. Red shows similar data for the neonatology only.
Outbreak investigations of nosocomial infections help to limit the healthcare burden of this increasing threat. In this study, we aimed to extend the knowledge on E. hormaechei gathered by the above-mentioned patient reports via implanting bacterial WGS. Our results provided extended information on the phylogenetic relationship and presence of AMR genes, MGEs and plasmids. The detection of class C β-lactamase genes in all isolates correlated with the MDR phenotype, which led to their classification as 2MRGN NeoPaed. However, despite 9 of 10 strains carrying the fosA gene, two isolates exhibited a reduced fosfomycin MIC. The reason for the lower Fos MIC in isolate VA41244 could be lack of expression of a functional FosA protein under the conditions used for AST [54]. The identification of AMR genes in bacterial WGS data is a very valuable approach to investigating MDR strains and the possible transfer of these genes to other bacteria in a clinical setting [55]. The transfer of AMR genes within a species but also across species boundaries can occur via horizontal gene transfer (HGT). The main routes of HGT in bacteria are conjugation, transduction, and the uptake of free DNA (natural transformation). The transfer of AMR genes via HGT is much more efficient if these genes are linked to MGEs, e.g., plasmids, phages or IS elements [56]. To investigate the potential of our strains to transfer AMR through HGT, we reconstructed their plasmids from short-read data. With this approach, we were able to assemble two plasmids of ~2.5 kb and ~4.7 kb length to circular contigs for the majority of the isolates. The reconstruction of the other plasmids was less efficient and would have required long-read sequencing techniques for improvement [57]. Nonetheless, we were able to assign the contigs to their plasmid origin with none of the AMR genes present on these fragments. Furthermore, because none of the AMR genes were linked to a contig harbouring an MGE, we considered all our E. hormaechei isolates to have a low potential to transfer these genes to other bacteria in a clinical setting.
5. Conclusions and Future Aspects
Our findings highlight the importance of implementing WGS as an effective approach to preventing and controlling bacterial infections in NICUs. The use of WGS has the capacity to revolutionise the way outbreak investigations are conducted in healthcare settings. It will improve the knowledge of bacterial ecology and uncover nosocomial transmission pathways and chance clusters. Furthermore, WGS provides pertinent clinical information regarding antimicrobial resistance and virulence genes that may not be detected by other testing methods.
Although WGS may eventually replace other molecular genomic methods as the main tool for molecular subtyping, it will continue to serve as a supplementary tool for traditional surveillance and epidemiological investigations, such as evaluating transmission points. While the robustness and scalability of WGS to accurately identify bacterial strains significantly improves patient care, several obstacles need to be addressed to routinely implement whole-genome sequencing (WGS) for outbreak investigations. These barriers include the turnaround time, accessibility, cost, and standardisation, as discussed by Sansom et al. [58]. In the future, WGS might emerge as the primary genomic surveillance tool with prospective potential [59], enabling the early identification of potential transmissions. This could contribute to a reduction in the morbidity and mortality rates associated with outbreaks.
References
- Flannery, D.D.; Chiotos, K.; Gerber, J.S.; Puopolo, K.M. Neonatal multidrug-resistant gram-negative infection: Epidemiology, mechanisms of resistance, and management. Pediatr. Res. 2022, 91, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Glaser, M.A.; Hughes, L.M.; Jnah, A.; Newberry, D. Neonatal Sepsis: A Review of Pathophysiology and Current Management Strategies. Adv. Neonatal Care 2021, 21, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wendt, C.; von Baum, H.; Kaase, M.; Meyer, E.; Suger-Wiedeck, H.; Ruscher, C. Hygienemaßnahmen bei Infektionen oder Besiedlung mit multiresistenten gramnegativen Stäbchen. Bundesgesundheitsblatt-Gesundheitsforschung-Gesundheitsschutz 2012, 55, 1311–1354. [Google Scholar] [CrossRef]
- Simon, A.; Christoph, J.; Geffers, C.; Hentschel, J.; Jürs, U.; Kramer, A.; Laux, R.A.; Müller, A.; Wendt, C. Recommendation for the prevention of nosocomial infections in neonatal intensive care patients with a birth weight less than 1500 g. Report by the Committee of Hospital Hygiene and Infection Prevention of the Robert Koch Institute. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2007, 50, 1265–1303. [Google Scholar] [CrossRef]
- Graham, P.L., 3rd; Begg, M.D.; Larson, E.; Della-Latta, P.; Allen, A.; Saiman, L. Risk factors for late onset gram-negative sepsis in low birth weight infants hospitalized in the neonatal intensive care unit. Pediatr. Infect. Dis. J. 2006, 25, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Parm, U.; Metsvaht, T.; Sepp, E.; Ilmoja, M.L.; Pisarev, H.; Pauskar, M.; Lutsar, I. Risk factors associated with gut and nasopharyngeal colonization by common Gram-negative species and yeasts in neonatal intensive care units patients. Early Hum. Dev. 2011, 87, 391–399. [Google Scholar] [CrossRef]
- Shakil, S.; Ali, S.Z.; Akram, M.; Ali, S.M.; Khan, A.U. Risk factors for extended-spectrum beta-lactamase producing Escherichia coli and Klebsiella pneumoniae acquisition in a neonatal intensive care unit. J. Trop. Pediatr. 2010, 56, 90–96. [Google Scholar] [CrossRef]
- Anderson, B.; Nicholas, S.; Sprague, B.; Campos, J.; Short, B.; Singh, N. Molecular and descriptive epidemiologyof multidrug-resistant Enterobacteriaceae in hospitalized infants. Infect. Control Hosp. Epidemiol. 2008, 29, 250–255. [Google Scholar] [CrossRef]
- Graham, P.L., 3rd; Della-Latta, P.; Wu, F.; Zhou, J.; Saiman, L. The gastrointestinal tract serves as the reservoir for Gram-negative pathogens in very low birth weight infants. Pediatr. Infect. Dis. J. 2007, 26, 1153–1156. [Google Scholar] [CrossRef]
- Das, P.; Singh, A.K.; Pal, T.; Dasgupta, S.; Ramamurthy, T.; Basu, S. Colonization of the gut with Gram-negative bacilli, its association with neonatal sepsis and its clinical relevance in a developing country. J. Med. Microbiol. 2011, 60, 1651–1660. [Google Scholar] [CrossRef]
- Parm, Ü.; Metsvaht, T.; Sepp, E.; Ilmoja, M.L.; Pisarev, H.; Pauskar, M.; Lutsar, I. Mucosal surveillance cultures in predicting Gram-negative late-onset sepsis in neonatal intensive care units. J. Hosp. Infect. 2011, 78, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Saiman, L.; Zhou, J.; Della-Latta, P.; Jia, H.; Graham, P.L., 3rd. Concordance of Gastrointestinal Tract Colonization and Subsequent Bloodstream Infections with Gram-negative Bacilli in Very Low Birth Weight Infants in the Neonatal Intensive Care Unit. Pediatr. Infect. Dis. J. 2010, 29, 831–835. [Google Scholar] [CrossRef]
- Bär, A.; Schmitt-Grohé, S.; Held, J.; Lubig, J.; Hanslik, G.; Fahlbusch, F.B.; Reutter, H.; Woelfle, J.; van der Donk, A.; Schleier, M.; et al. Evaluating the Use of Neonatal Colonization Screening for Empiric Antibiotic Therapy of Sepsis and Pneumonia. Antibiotics 2023, 12, 189. [Google Scholar] [CrossRef] [PubMed]
- Baier, C.; Pirr, S.; Ziesing, S.; Ebadi, E.; Hansen, G.; Bohnhorst, B.; Bange, F.C. Prospective surveillance of bacterial colonization and primary sepsis: Findings of a tertiary neonatal intensive and intermediate care unit. J. Hosp. Infect. 2019, 102, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Schuurmans, J.; Ariti, C.; Borgundvaag, E.; Charles, K.; Badjo, C.; Clezy, K.; Evens, E.; Senat-Delva, R.; Berthet, M.; et al. Rectal screening displays high negative predictive value for bloodstream infection with (ESBL-producing) Gram-negative bacteria in neonates with suspected sepsis in a low-resource setting neonatal care unit. J. Glob. Antimicrob. Resist. 2020, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Feng, Y.; Zong, Z. Precise Species Identification for Enterobacter: A Genome Sequence-Based Study with Reporting of Two Novel Species, Enterobacter quasiroggenkampii sp. nov. and Enterobacter quasimori sp. nov. mSystems 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Pestourie, N.; Garnier, F.; Barraud, O.; Bedu, A.; Ploy, M.C.; Mounier, M. Outbreak of AmpC β-lactamase-hyper-producing Enterobacter cloacae in a neonatal intensive care unit in a French teaching hospital. Am. J. Infect. Control 2014, 42, 456–458. [Google Scholar] [CrossRef]
- Wendel, A.F.; Peter, D.; Mattner, F.; Weiss, M.; Hoppenz, M.; Wolf, S.; Bader, B.; Peter, S.; Liese, J. Surveillance of Enterobacter cloacae complex colonization and comparative analysis of different typing methods on a neonatal intensive care unit in Germany. Antimicrob. Resist. Infect. Control 2022, 11, 54. [Google Scholar] [CrossRef]
- Martineau, C.; Li, X.; Lalancette, C.; Perreault, T.; Fournier, E.; Tremblay, J.; Gonzales, M.; Yergeau, É.; Quach, C. Serratia marcescens Outbreak in a Neonatal Intensive Care Unit: New Insights from Next-Generation Sequencing Applications. J. Clin. Microbiol. 2018, 56, e00235-18. [Google Scholar] [CrossRef]
- Fleisch, F.; Zimmermann-Baer, U.; Zbinden, R.; Bischoff, G.; Arlettaz, R.; Waldvogel, K.; Nadal, D.; Ruef, C. Three consecutive outbreaks of Serratia marcescens in a neonatal intensive care unit. Clin. Infect. Dis. 2002, 34, 767–773. [Google Scholar] [CrossRef]
- Montagnani, C.; Cocchi, P.; Lega, L.; Campana, S.; Biermann, K.P.; Braggion, C.; Pecile, P.; Chiappini, E.; de Martino, M.; Galli, L. Serratia marcescens outbreak in a neonatal intensive care unit: Crucial role of implementing hand hygiene among external consultants. BMC Infect. Dis. 2015, 15, 11. [Google Scholar] [CrossRef]
- Dawczynski, K.; Proquitté, H.; Roedel, J.; Edel, B.; Pfeifer, Y.; Hoyer, H.; Dobermann, H.; Hagel, S.; Pletz, M.W. Intensified colonisation screening according to the recommendations of the German Commission for Hospital Hygiene and Infectious Diseases Prevention (KRINKO): Identification and containment of a Serratia marcescens outbreak in the neonatal intensive care unit, Jena, Germany, 2013–2014. Infection 2016, 44, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Bekal, S.; Berry, C.; Reimer, A.R.; Van Domselaar, G.; Beaudry, G.; Fournier, E.; Doualla-Bell, F.; Levac, E.; Gaulin, C.; Ramsay, D.; et al. Usefulness of High-Quality Core Genome Single-Nucleotide Variant Analysis for Subtyping the Highly Clonal and the Most Prevalent Salmonella enterica Serovar Heidelberg Clone in the Context of Outbreak Investigations. J. Clin. Microbiol. 2016, 54, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; SenGupta, D.J.; Cummings, L.A.; Land, T.A.; Hoogestraat, D.R.; Cookson, B.T. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J. Clin. Microbiol. 2015, 53, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, M.A.; Ozer, E.A.; Hauser, A.R. Utility of Whole-Genome Sequencing in Characterizing Acinetobacter Epidemiology and Analyzing Hospital Outbreaks. J. Clin. Microbiol. 2016, 54, 593–612. [Google Scholar] [CrossRef] [PubMed]
- Shogan, B.D.; Smith, D.P.; Packman, A.I.; Kelley, S.T.; Landon, E.M.; Bhangar, S.; Vora, G.J.; Jones, R.M.; Keegan, K.; Stephens, B.; et al. The Hospital Microbiome Project: Meeting report for the 2nd Hospital Microbiome Project, Chicago, USA, January 15(th), 2013. Stand Genom. Sci. 2013, 8, 571–579. [Google Scholar] [CrossRef]
- Hewitt, K.M.; Mannino, F.L.; Gonzalez, A.; Chase, J.H.; Caporaso, J.G.; Knight, R.; Kelley, S.T. Bacterial diversity in two Neonatal Intensive Care Units (NICUs). PLoS ONE 2013, 8, e54703. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A.; Underwood, M.A. Surface microbes in the neonatal intensive care unit: Changes with routine cleaning and over time. J. Clin. Microbiol. 2013, 51, 2617–2624. [Google Scholar] [CrossRef]
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 153. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Sutton, G.G.; Brinkac, L.M.; Clarke, T.H.; Fouts, D.E. Enterobacterhormaechei subsp. hoffmannii subsp. nov., Enterobacter hormaechei subsp. xiangfangensis comb. nov., Enterobacter roggenkampii sp. nov., and Enterobacter muelleri is a later heterotypic synonym of Enterobacter asburiae based on computational analysis of sequenced Enterobacter genomes. F1000Research 2018, 7, 521. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Ahmed, S.A.; Davenport, K.W.; Flynn, M.C.; Lo, C.C.; Chain, P.S.G. Standardized phylogenetic and molecular evolutionary analysis applied to species across the microbial tree of life. Sci. Rep. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.; Zhu, H.; Guan, Y.; Lam, T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2016, 8, 28–36. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 15 November 2022).
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Schmartz, G.P.; Hartung, A.; Hirsch, P.; Kern, F.; Fehlmann, T.; Müller, R.; Keller, A. PLSDB: Advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022, 50, D273–D278. [Google Scholar] [CrossRef]
- Pavlovic, M.; Konrad, R.; Iwobi, A.N.; Sing, A.; Busch, U.; Huber, I. A dual approach employing MALDI-TOF MS and real-time PCR for fast species identification within the Enterobacter cloacae complex. FEMS Microbiol. Lett. 2012, 328, 46–53. [Google Scholar] [CrossRef]
- Boerlin, P.; Reid-Smith, R.J. Antimicrobial resistance: Its emergence and transmission. Anim. Health Res. Rev. 2008, 9, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Dunne, W.M., Jr.; Westblade, L.F.; Ford, B. Next-generation and whole-genome sequencing in the diagnostic clinical microbiology laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 2011, 6, e22751. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R.L.; Pouseele, H.; Chen, J.C.; Strockbine, N.A.; Carleton, H.A. Implementation of Whole Genome Sequencing (WGS) for Identification and Characterization of Shiga Toxin-Producing Escherichia coli (STEC) in the United States. Front. Microbiol. 2016, 7, 766. [Google Scholar] [CrossRef] [PubMed]
- Godmer, A.; Benzerara, Y.; Normand, A.C.; Veziris, N.; Gallah, S.; Eckert, C.; Morand, P.; Piarroux, R.; Aubry, A. Revisiting Species Identification within the Enterobacter cloacae Complex by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. Microbiol. Spectr. 2021, 9, e0066121. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, C.M.; Steigerwalt, A.G.; Hill, B.C.; Farmer, J.J., 3rd; Fanning, G.R.; Brenner, D.J. Enterobacter hormaechei, a new species of the family Enterobacteriaceae formerly known as enteric group 75. J. Clin. Microbiol. 1989, 27, 2046–2049. [Google Scholar] [CrossRef] [PubMed]
- Dyabi, F.Z.; Bennaoui, F.; El Idrissi Slitine, N.; Soraa, N.; Maoulainine, F.M.R. Enterobacter Hormaechei: New Neonatal Infection in Morocco. Open Infect. Dis. J. 2018, 10, 4. [Google Scholar] [CrossRef]
- da Silva, C.L.; Miranda, L.E.; Moreira, B.M.; Rebello, D.; Carson, L.A.; Kellum, M.E.; de Almeida, M.C.; Sampaio, J.L.; O’Hara, C.M. Enterobacter hormaechei bloodstream infection at three neonatal intensive care units in Brazil. Pediatr. Infect. Dis. J. 2002, 21, 175–177. [Google Scholar] [CrossRef]
- Girlich, D.; Ouzani, S.; Emeraud, C.; Gauthier, L.; Bonnin, R.A.; Le Sache, N.; Mokhtari, M.; Langlois, I.; Begasse, C.; Arangia, N.; et al. Uncovering the novel Enterobacter cloacae complex species responsible for septic shock deaths in newborns: A cohort study. Lancet Microbe 2021, 2, e536–e544. [Google Scholar] [CrossRef]
- Lax, S.; Sangwan, N.; Smith, D.; Larsen, P.; Handley, K.M.; Richardson, M.; Guyton, K.; Krezalek, M.; Shogan, B.D.; Defazio, J.; et al. Bacterial colonization and succession in a newly opened hospital. Sci. Transl. Med. 2017, 9, eaah6500. [Google Scholar] [CrossRef] [PubMed]
- Capone, K.A.; Dowd, S.E.; Stamatas, G.N.; Nikolovski, J. Diversity of the human skin microbiome early in life. J. Investig. Dermatol. 2011, 131, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Elliott, Z.S.; Barry, K.E.; Cox, H.L.; Stoesser, N.; Carroll, J.; Vegesana, K.; Kotay, S.; Sheppard, A.E.; Wailan, A.; Crook, D.W.; et al. The Role of fosA in Challenges with Fosfomycin Susceptibility Testing of Multispecies Klebsiella pneumoniae Carbapenemase-Producing Clinical Isolates. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef]
- Köser, C.U.; Ellington, M.J.; Peacock, S.J. Whole-genome sequencing to control antimicrobial resistance. Trends Genet. 2014, 30, 401–407. [Google Scholar] [CrossRef]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef]
- Berbers, B.; Ceyssens, P.J.; Bogaerts, P.; Vanneste, K.; Roosens, N.H.C.; Marchal, K.; De Keersmaecker, S.C.J. Development of an NGS-Based Workflow for Improved Monitoring of Circulating Plasmids in Support of Risk Assessment of Antimicrobial Resistance Gene Dissemination. Antibiotics 2020, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Sansom, S.E.; Logan, L.K.; Green, S.J.; Moore, N.M.; Hayden, M.K. Whole-genome sequencing for neonatal intensive care unit outbreak investigations: Insights and lessons learned. Antimicrob. Steward. Healthc. Epidemiol. 2021, 1, e2. [Google Scholar] [CrossRef]
- Ward, D.V.; Hoss, A.G.; Kolde, R.; van Aggelen, H.C.; Loving, J.; Smith, S.A.; Mack, D.A.; Kathirvel, R.; Halperin, J.A.; Buell, D.J.; et al. Integration of genomic and clinical data augments surveillance of healthcare-acquired infections. Infect. Control Hosp. Epidemiol. 2019, 40, 649–655. [Google Scholar] [CrossRef] [PubMed]

