


1. Introduction
Hyperlipidemia is defined as elevated levels of triglycerides and/or any of the following lipoproteins: very-low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs), or high-density lipoproteins (HDLs). Hyperlipidemia expression is replaced by dyslipidemia as increasing HDL levels is a good sign [1]. Dyslipidemia is classified into familial (primary) dyslipidemia, which is caused by genetic disorders, and acquired (secondary) dyslipidemia, caused by the progression or signs of some diseases like diabetes, kidney disorder, and hypothyroidism [2,3]. Also, hyperlipidemia can increase the risk of developing some medical conditions like bladder cancer and coronary artery diseases [4]. Some cases report that dyslipidemia appears in overweight pediatrics. As a risk factor for management, triglycerides, total cholesterol, very-low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs) are periodically analyzed for patients to prevent atherosclerosis [3]. The first treatment for controlling dyslipidemia is lifestyle management, e.g., decreasing fat and high-cholesterol diet intake. Several drug categories are used to manage the level of serum lipid. The first group is bile acid binders such as cholestyramine, colesovelam, and colestipol. The second group is fibrates, e.g., fenofibrate and gemfibrozil, which stimulate the cells’ fatty acid uptake, convert it to acyl-CoA derivatives, and then catabolize it via oxidative pathways [5]. The third lipid-lowering group is cholesterol absorption inhibitors, e.g., ezetimibe, which significantly decreases the absorbed quantity of cholesterol. The fourth group is considered a supplement rather than a drug, which is omega-3 fatty acids that act by inhibiting VLDL synthesis. The fifth and most common group used for managing dyslipidemia is the 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-COA) reductase inhibitors (statin). This group prevents the transformation of HMG-COA into mevalonate. The statin group contains simvastatin, pravastatin, atorvastatin, lovastatin, pitavastatin, and rosuvastatin. This medicine group is classified according to the biopharmaceutics classification system (BCS) as a class II drug characterized by low solubility and high permeability. Thus, it causes low bioavailability in this group. In addition, it shows poor acid stability and is highly affected by the first-pass effect. Thus, rosuvastatin exhibits a low bioavailability of about 20% [3].
Rosuvastatin calcium (RSV) is a synthetic lipid-lowering agent, chemically known as (3R,5S,6E)-7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl])-3,5-dihydroxyhept-6-enoic acid hemicalcium salt [6]. Rosuvastatin, among other statins, is called a “super-statin,” causing a greater reduction in LDL than other statins of the same strength [7,8,9]. Several recent approaches have been published to improve rosuvastatin’s bioavailability using different mechanisms. Elsayed and his coworkers prepared forming nanoparticles in situ with the aid of Tween 80 and cetyl alcohol and filled in delayed-release capsules that improved the dissolution rate and bioavailability [10]. In addition, reducing the particle size of rosuvastatin using a wet milling technique by adding PVP 10% as a stabilizer enhanced its dissolution behavior to release 72% after 1 h [7]. Furthermore, the development of pullulan-based tablets containing flexible chitosomes of rosuvastatin calcium improved relative bioavailability by 30% to 36% compared to marketed drugs and pure rosuvastatin tablets [3]. Also, using caffeine and Soluplus® to develop hydrotropic and micellar solubilization is another approach to directly compress rosuvastatin with improved bioavailability [11]. In addition, incorporating RSV into carboxylate cross-linked cyclodextrins improved its bioavailability [12]. Recently, González and his coworkers improved the bioavailability of RSV via its conversion to an amorphous form with a specific excipient to accelerate its dissolution onset by more than 90% in 10 min [13]. Also, RSV was incorporated with glimepiride in 3D-printed polypills formulated in a curcuma oil-based self-nanoemulsifying drug delivery system to treat patients with dyslipidemia and metabolic syndrome [14]. In addition, trials to formulate RSV as orodispersable films were performed with sophisticated and multi-stage procedures. The films produced by this work were evaluated for pharmacokinetic parameters, not for anti-dyslipidemic activity, as declared by our study [15].
Among other routes of administration, the oral route proved to yield optimum patient acceptability, as it is non-invasive and self-administered. There are many dosage forms administered orally. Some are wholly ingested; others can be chewed, dissolved in a specific solvent before taking, or adhered to the tongue or buccal cavity. Taking the dose via ingestion forced the active pharmaceutical ingredients into some challenges, like facing a low pH medium in the stomach, as many active pharmaceutical ingredients are unstable in acidic media. Also, some drugs are affected by first-pass metabolism prior to absorption. The relatively low bioavailability of some drugs after oral ingestion creates many challenges for developers to find a way to protect the drugs labile to these situations, like formulating them in delayed-release dosage forms.
Fast-dissolving films are considered a new oral dosage form that offers immediate action with a reasonable degree of protection from stomach acidity and the first-pass effect, as the dissolution and absorption phases are carried out in the oral cavity. It is a waterless dosage form and provides the action with a fast beginning. Fast-dissolving films, among other dosage forms, are highly accepted by pediatric and elderly patients due to their ease of use. Recently, many researchers published new polymer-based fast-dissolving films, e.g., fluoxetine [16], metoclopramide [17], lamotrigine [18], ondansetron hydrochloride [19], olanzapine [20], and tenoxicam [21]. Fast-dissolving films can be prepared using various techniques, such as solvent casting, characterized by combining the polymer solution with the plasticizer and drug solution, mixing, degassing, pouring into a suitable dish, and heating to evaporate the solvent [21]. Other methods include hot melt extrusion, semisolid casting, solid dispersion extrusion, and rolling [22]. A new approach was recently applied for preparing fast-dissolving dosage forms using a spinning agent that freely dissolves the drug of interest and is mixed with the polymeric solution [23].
Therefore, this work aimed to develop rosuvastatin calcium as a fast-dissolving film to be rapidly dissolved and absorbed in the buccal cavity. This approach helped to avoid the first-pass effect, protect the drug from degradation by stomach acidity, and subsequently improve the bioavailability of RSV.
2. Materials and Methods
2.1. Materials
Polyethylene glycol 400 (PEG 400) and rosuvastatin calcium were gifted from Egyptian International Pharmaceutical Industries Co. (10th of Ramadan, Egypt). Future Pharmaceutical Industries (Badr City, Egypt) provided hydroxypropyl methylcellulose (HPMC), viscosity 4000 cp, as a gift. Acetonitrile, ortho-phosphoric acid, methanol, and potassium dihydrogen phosphate were purchased from Merck (Darmstadt, Germany). Mannitol, Sorbitol, and Poloxamer 407 (P407, MW 40000) were obtained as a gift from Medical Union Pharmaceuticals (Ismailia, Egypt).
2.2. Methods
3. Results and Discussion
In the current work, optimized RSV-FDFs were developed by tailoring a polymeric matrix with the aid of hydroxypropyl methylcellulose and the plasticizing effect of glycerin. The formulation factors were investigated to determine their effects on the quality of the prepared FDFs and predict the optimum levels that produce the optimized formulation with the desired quality attributes. This optimized RSV-FDF was evaluated for its pharmacokinetic behavior and anti-dyslipidemic activity.
3.1. Formulation and Evaluation of RSV-FDFs
The evaluation of the prepared films’ physicomechanical properties is provided in . The films were found to be soft, clear, thin, and colorless, with no bubbles entrapped, and there were no issues during removal from the dish or the cutting procedures. The film clarity demonstrates that the drug was already soluble in the film polymer and thus supports the results of in vitro dissolution, which demonstrated immediate release after the disintegration of the film.
To assess the uniformity of the RSV distribution within the formula, five different places in each formula were analyzed to determine the drug content in each formula, and the results show that the drug was distributed uniformly throughout the films and within the accepted and required compendial specifications, with an RSD% of less than 10%. Also, the uniformity of weight in all films yielded an acceptable RSD%.
As the normal pH range of saliva lies between 6.2 and 7.6 [21], any acidic or basic pH distortion from normal salivary pH will cause irritation and patient noncompliance with the treatment protocol. All prepared films revealed a pH range of 6.5–6.62, ensuring no irritation to the oral cavity upon administration.
3.2. Optimization of RSV-FDFs
Ten experimental runs were suggested by a 22+star central composite design to demonstrate the effect of the following independent variables: polymer percentage (X1) from 1 to 3% and plasticizer percentage (X2) from 1 to 2% on the disintegration time (Y1), and thickness (Y2) and folding endurance (Y3) of the prepared films.
3.3. In Vivo Pharmacokinetics Evaluation
The plasma concentration-time curve acquired after dosing male Wistar rats with 20mg/kg rosuvastatin from the commercial product (M) and fast-dissolving film formula (F) is demonstrated in Figure 3. The pharmacokinetic parameters were calculated using WinNonLin® 8.2 software (Princeton, NJ, USA) and are listed in . The absorption was monitored for a period of 72 h. The differences in Tmax were used to evaluate these data. The RSV-FDF formula showed faster release, which was revealed in the reduction of the Tmax, and the extent of the absorbed drug improved, which appeared as a higher Cmax for the RSV-FDF (1.540 ± 0.044 µg/mL) and as 0.940 ± 0.017 µg/mL for the marketed product. In comparison to the commercial formula, relative bioavailability improved by 32.5%.
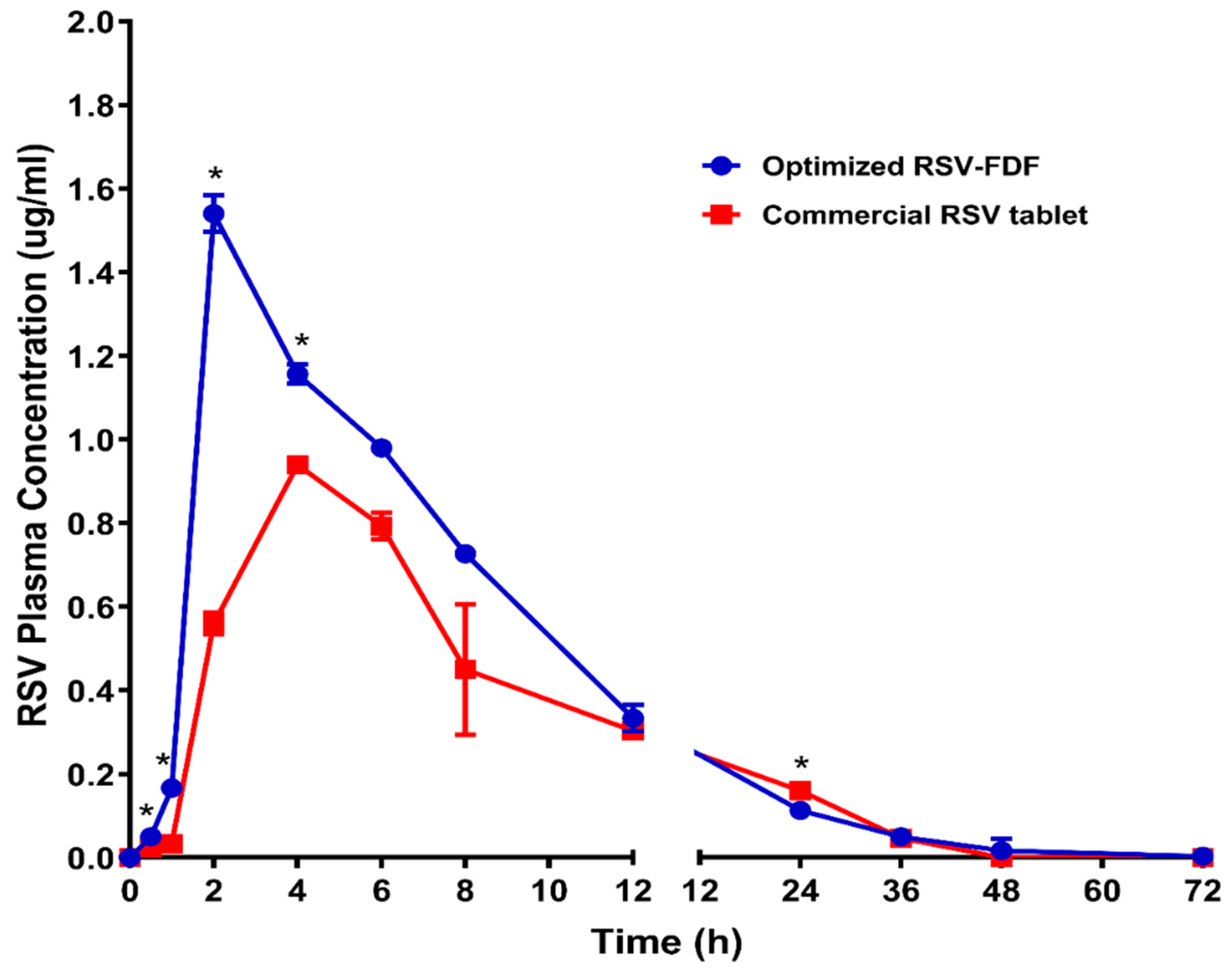
Figure 3. Rosuvastatin calcium plasma time concentration curves after administration of optimized RSV-FDFs and commercial RSV tablets. Data represent the mean value ± standard deviation (SD). * Significant at p < 0.05.
Furthermore, the multiple t-test using the Holm–Sidak method revealed that Cmax, AUC0–t, AUC0–inf, and the clearance (Cl) showed significant differences between the optimized RSV-FDF and the commercial oral tablet, with p-values of 0.000025, 0.002059, 0.001063, and 0.002239, respectively.
The improvement in Tmax directly referred to the origin of the formula of the RSV-FDF containing the rosuvastatin in the dissolved state, so there was only 1 min for average disintegration with no dissolution time required. At the same time, the marketed product needed more time for the disintegration and dissolution stages. The improvement in the RSV-FDF is also referred to as bypassing the first pass effect and protecting rosuvastatin from degradation in acidic media.
3.4. In Vivo Pharmacodynamics Evaluation
The hypolipidemic activity of rosuvastatin was referenced to prevent the synthesis of mevalonic acid from its precursor HMG-COA by inhibiting the enzyme HMG COA reductase, which decreases the lipid profile.
To study the efficiency of the RSV-FDF on the lipid profile (triglycerides, total cholesterol, LDLs, VLDLs, and HDLs) in rats with induced hyperlipidemia, Poloxamer 407 was administered to male Wistar rats 24 h prior to the experiment by to induce hyperlipidemia; then, the rats categorized into three groups (n = 3): negative control (C), commercial product (M), and RSV-FDF (O). Then, zero time samples were collected from each group to define the baseline for each parameter and the efficacy was determined for the O group, which showed a decrease in total cholesterol of 68.1% after 6 h from the zero time point, which is a significant difference in comparison to the M group, as the total cholesterol was reduced by 58.2% and the reduction In total cholesterol persisted for 24 h. The level of triglycerides also decreased by 56.4% for the O group, whereas the M group’s level of triglycerides was reduced by 37.6%. Also, for the LDLs, the O group showed better performance, as after 6 h, the level of LDLs in the O group decreased by about 60.6% compared to 50% in the marketed group. The following Figure 4 shows the graphical representation of the different parameters of the lipid profiles in the three groups.
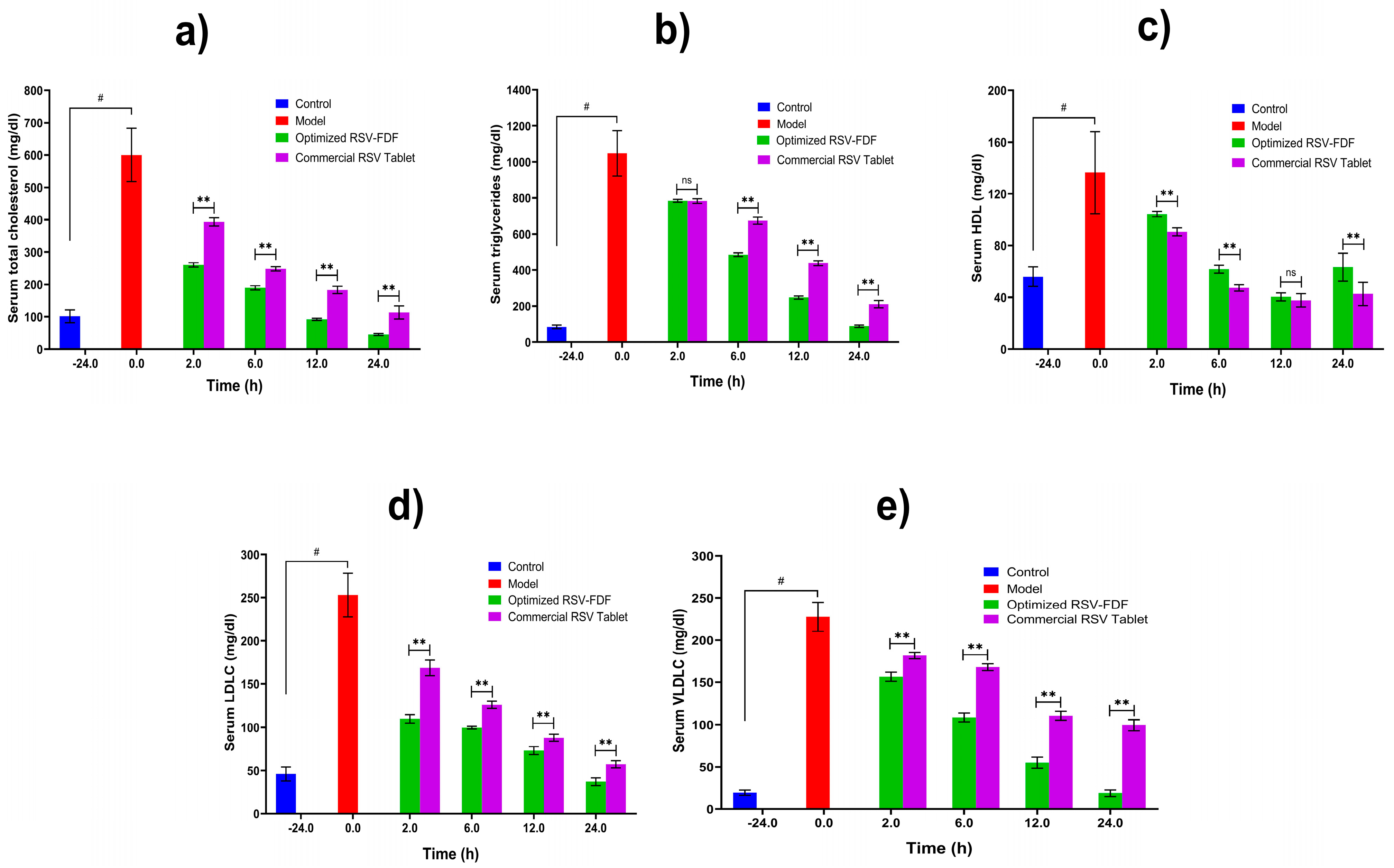
Figure 4. Lipid profiles of induced hyperlipidemic rats after single-dose administration of optimized RSV-FDFs and commercial RSV tablets. Data are presented as mean ± SD (n = 3). Note: # denotes a significant difference between normal and model at p < 0.05, ** denotes a significant difference between the optimized RSV-FDF and the commercial RSV tablets at p < 0.01, and ns denotes a non-significant difference. (a) the total serum cholesterol level in mg/dl, (b) the total serum triglyceride level mg/dl, (c) the serum HDL level mg/dl, (d) the serum LDL level mg/dl, and (e) the serum VLDL level mg/dl.
4. Conclusions
The prepared fast-dissolving film formula containing rosuvastatin (RSV-FDF) yielded acceptable results regarding in vitro characterization and evaluation. The pharmacokinetics supported these findings, which revealed a significant improvement in relative bioavailability of 32.5%. The pharmacodynamic experiments also showed significant improvements for RSV-FDF compared to the commercial rosuvastatin tablets of a 50% reduction in triglyceride levels and a 21% reduction in LDL values. Therefore, the RSV-FDF can be considered a promising substitute for commercial tablets, although additional studies in humans and extra stability determinations should be carried out in the future.
References
- Mosca, S.; Araújo, G.; Costa, V.; Correia, J.; Bandeira, A.; Martins, E.; Mansilha, H.; Tavares, M.; Coelho, M.P. Dyslipidemia Diagnosis and Treatment: Risk Stratification in Children and Adolescents. J. Nutr. Metab. 2022, 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Peng, H.; Chen, X.; Wu, X.; Wang, B. Hyperlipidemia and hypothyroidism. Clin. Chim. Acta 2022, 527, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.; Elimam, H.; Alrifai, A.O.; Nadhrah, H.M.; Masoudi, L.Y.; Sairafi, W.O.; M.El-Say, K. Rosuvastatin lyophilized tablets loaded with flexible chitosomes for improved drug bioavailability, anti-hyperlipidemic and anti-oxidant activity. Int. J. Pharm. 2020, 558, 119791. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-J.; Lin, K.-H.; Wen, Y.-C.; Fan, Y.-C.; Tsai, P.-S.; Huang, C.-J. Increased risk of bladder cancer in young adult men with hyperlipidemia: A population-based cohort study. Medicine 2021, 100, e28125. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H.; Fager, G. Pharmacodynamics and Pharmacokinetics of the HMG-CoA Reductase Inhibitors. Clin. Pharmacokinet. 1997, 32, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Alshora, D.H.; Ibrahim, M.A.; Elzayat, E.; Almeanazel, O.T.; Alanazi, F. Rosuvastatin calcium nanoparticles: Improving bioavailability by formulation and stabilization codesign. PLoS ONE 2018, 13, e0200218. [Google Scholar] [CrossRef]
- Chizner, M.A.; Duvall, W.L. Highlights of prescribing information crestor (rosuvastatin calcium). Cardiovasc. Rev. Rep. 2003, 24, 591. [Google Scholar]
- Scott, L.J.; Curran, M.P.; Figgitt, D.P. Rosuvastatin. Am. J. Cardiovasc. Drugs 2004, 4, 117–138. [Google Scholar] [CrossRef]
- Elsayed, I.; El-Dahmy, R.M.; Elshafeey, A.H.; El Gawad, N.A.A.; El Gazayerly, O.N. Tripling the bioavailability of rosuvastatin calcium through development and optimization of an In-Situ forming nanovesicular system. Pharmaceutics 2019, 11, 275. [Google Scholar] [CrossRef]
- Butt, S.; Hasan, S.M.F.; Hassan, M.M.; Alkharfy, K.M.; Neau, S.H. Directly compressed rosuvastatin calcium tablets that offer hydrotropic and micellar solubilization for improved dissolution rate and extent of drug release. Saudi Pharm. J. 2019, 27, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.M.; Mortada, S.M.; Sallam, M.A. Carboxylate cross-linked cyclodextrin: A nanoporous scaffold for enhancement of rosuvastatin oral bioavailability. Eur. J. Pharm. Sci. 2018, 111, 1–12. [Google Scholar] [CrossRef] [PubMed]
- González, R.; Peña, M.; Torres, N.S.; Torrado, G. Design, development, and characterization of amorphous rosuvastatin calcium tablets. PLoS ONE 2022, 17, e0265263. [Google Scholar] [CrossRef] [PubMed]
- El-Say, K.M.; Felimban, R.; Tayeb, H.H.; Chaudhary, A.G.; Omar, A.M.; Rizg, W.Y.; Alnadwi, F.H.; I Abd-Allah, F.; Ahmed, T. Pairing 3D-Printing with Nanotechnology to Manage Metabolic Syndrome. Int. J. Nanomed. 2022, 17, 1783–1801. [Google Scholar] [CrossRef] [PubMed]
- Zaki, R.M.; Alfadhel, M.; Seshadri, V.D.; Albagami, F.; Alrobaian, M.; Tawati, S.M.; Warsi, M.H.; Almurshedi, A.S. Fabrication and characterization of orodispersible films loaded with solid dispersion to enhance Rosuvastatin calcium bioavailability. Saudi Pharm. J. 2023, 31, 135–146. [Google Scholar] [CrossRef]
- Rédai, E.-M.; Antonoaea, P.; Todoran, N.; Vlad, R.A.; Bîrsan, M.; Tătaru, A.; Ciurba, A. Development and evaluation of fluoxetine fast dissolving films: An alternative for noncompliance in pediatric patients. Processes 2021, 9, 778. [Google Scholar] [CrossRef]
- Reveny, J.; Tanuwijaya, J.; Remalya, A. Formulation of Orally Dissolving Film (ODF) Metoclopramide Using Hydroxy Propyl Methyl Cellulose and Polyvinyl Alcohol with Solvent Casting Method. Int. J. ChemTech Res. 2017, 10, 316–321. [Google Scholar]
- Hamza, M. Development and Evaluation of Orodispersible Films of Lamotrigine: Hydroxypropyl Β Cyclodextrin Inclusion Complex. Al-Azhar J. Pharm. Sci. 2017, 56, 31–46. [Google Scholar] [CrossRef]
- Koland, M.; Sandeep, V.; Charyulu, N. Fast Dissolving Sublingual Films of Ondansetron Hydrochloride: Effect of Additives on in vitro Drug Release and Mucosal Permeation. J. Young- Pharm. 2010, 2, 216–222. [Google Scholar] [CrossRef]
- Cho, H.-W.; Baek, S.-H.; Lee, B.-J.; Jin, H.-E. Orodispersible polymer films with the poorly water-soluble drug, olanzapine: Hot-Melt pneumatic extrusion for single-process 3D printing. Pharmaceutics 2020, 12, 692. [Google Scholar] [CrossRef]
- Abdulelah, F.M.; Abdulbaqi, M.R. Fast dissolving film nanocrystal (FDFN) preparation as a new trend for solubility enhancement of poorly soluble class ii drug tenoxicam. AVFT–Arch. Venez. Farmacol. Ter. 2021, 40, 333–339. [Google Scholar]
- Hoffmann, E.M.; Breitenbach, A.; Breitkreutz, J.; Pharm, D. Advances in orodispersible films for drug delivery. Expert Opin. Drug Deliv. 2011, 8, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Zhou, F.; Williams, G.R. Developing and scaling up fast-dissolving electrospun formulations based on poly(vinylpyrrolidone) and ketoprofen. J. Drug Deliv. Sci. Technol. 2020, 61, 102138. [Google Scholar] [CrossRef]
- Adrover, A.; Varani, G.; Paolicelli, P.; Petralito, S.; Di Muzio, L.; Casadei, M.A.; Tho, I. Experimental and modeling study of drug release from HPMC-based erodible oral thin films. Pharmaceutics 2018, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Hosny, K.M.; El-Say, K.M.; Ahmed, O.A. Optimized sildenafil citrate fast orodissolvable film: A promising formula for overcoming the barriers hindering erectile dysfunction treatment. Drug Deliv. 2014, 23, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Rathod, S.; Phansekar, M.; Bhagwan, A.; Surve, G. A Review on mouth dissolving tablets. Indian Drugs 2013, 50, 5–14. [Google Scholar] [CrossRef]
- Maher, E.M.; Ali, A.M.A.; Salem, H.F.; Abdelrahman, A.A. In vitro/in vivo evaluation of an optimized fast dissolving oral film containing olanzapine co-amorphous dispersion with selected carboxylic acids. Drug Deliv. 2016, 23, 3088–3100. [Google Scholar] [CrossRef]
- Wasilewska, K.; Winnicka, K. How to assess orodispersible film quality? A review of applied methods and their modifications. Acta Pharm. 2019, 69, 155–176. [Google Scholar] [CrossRef]
- Vishvakarma, P. Design and development of montelukast sodium fast dissolving films for better therapeutic efficacy. J. Chil. Chem. Soc. 2018, 63, 3988–3993. [Google Scholar] [CrossRef]
- Kumar, A.; Verma, R.; Jain, V. Formulation Development and Evaluation of Fast Dissolving Oral Film of Dolasetron Mesylate. Asian J. Pharm. Educ. Res. 2019, 8, 38. [Google Scholar] [CrossRef]
- Liew KBin Tan, Y.T.F.; Peh, K.K. Effect of polymer, plasticizer and filler on orally disintegrating film. Drug. Dev. Ind. Pharm. 2014, 40, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Elshafeey, A.H.; El-Dahmy, R.M. Formulation and development of oral fast-dissolving films loaded with nanosuspension to augment paroxetine bioavailability: In vitro characterization, ex vivo permeation, and pharmacokinetic evaluation in healthy human volunteers. Pharmaceutics 2021, 13, 1869. [Google Scholar] [CrossRef] [PubMed]
- Olechno, K.; Maciejewski, B.; Głowacz, K.; Lenik, J.; Ciosek-Skibińska, P.; Basa, A.; Winnicka, K. Orodispersible Films with Rupatadine Fumarate Enclosed in Ethylcellulose Microparticles as Drug Delivery Platform with Taste-Masking Effect. Materials 2022, 15, 2126. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y.; Lee, C.-J.; Lin, Y.-Y. Drug-polymer interaction affecting the mechanical properties, adhesion strength and release kinetics of piroxicam-loaded Eudragit E films plasticized with different plasticizers. J. Control. Release 1995, 33, 375–381. Available online: http://www.sciencedirect.com/science/article/pii/0168365994001098 (accessed on 23 October 2023). [CrossRef]
- Pichayakorn, W.; Suksaeree, J.; Boonme, P.; Amnuaikit, T.; Taweepreda, W.; Ritthidej, G.C. Deproteinized natural rubber film forming polymeric solutions for nicotine transdermal delivery. Pharm. Dev. Technol. 2011, 18, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Rujivipat, S.; Bodmeier, R. Moisture plasticization for enteric Eudragit® L30D-55-coated pellets prior to compression into tablets. Eur. J. Pharm. Biopharm. 2012, 81, 223–229. [Google Scholar] [CrossRef]
- Nesseem, D.I.; Eid, S.; El-Houseny, S. Development of novel transdermal self-adhesive films for tenoxicam, an anti-inflammatory drug. Life Sci. 2011, 89, 430–438. [Google Scholar] [CrossRef]
- Bhupinder, B.; Sarita, J. Formulation and evaluation of fast dissolving sublingual films of Rizatriptan Benzoate. Int. J. Drug. Dev. Res. 2012, 4, 133–143. [Google Scholar]
- Choi, M.-J.; Woo, M.R.; Choi, H.-G.; Jin, S.G. Effects of Polymers on the Drug Solubility and Dissolution Enhancement of Poorly Water-Soluble Rivaroxaban. Int. J. Mol. Sci. 2022, 23, 9491. [Google Scholar] [CrossRef]
- Al-Mogherah, A.I.; Ibrahim, M.A.; Hassan, M.A. Optimization and evaluation of venlafaxine hydrochloride fast dissolving oral films. Saudi Pharm. J. 2020, 28, 1374–1382. [Google Scholar] [CrossRef]
- Centkowska, K.; Ławrecka, E.; Sznitowska, M. Technology of orodispersible polymer films with micronized loratadine—influence of different drug loadings on film properties. Pharmaceutics 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Mankad, A.; Dutta, A. Methylphenidate Fast Dissolving Films: Development, Optimization Using Simplex Centroid Design and In Vitro Characterization. Turk. J. Pharm. Sci. 2022, 19, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Bharti, K.; Mittal, P.; Mishra, B. Formulation and characterization of fast dissolving oral films containing buspirone hydrochloride nanoparticles using design of experiment. J. Drug Deliv. Sci. Technol. 2018, 49, 420–432. [Google Scholar] [CrossRef]