


1. Introduction
Colorectal cancer (CRC) was ranked third and second in terms of incidence and mortality, respectively, accounting for 1.9 million of all new cancer cases and 900,000 deaths in 2020 worldwide. Notably, the incidence of CRC has shown a rising trend among individuals aged less than 50 years [1].
CRC neo-tumorigenesis is a complex and multistep process, linked to factors of oncogenesis such as oxidative stress (OS), chronic inflammation, reprogrammed glucose metabolism and diabetes mellitus (DM). In addition, defective signaling or changes in the expression of proteins regulating programmed cell death ‘’apoptosis’’ play crucial roles in the evolution of normal mucosa cells towards neoplastic transformations and the acquisition of new characteristics, among which evading cell death [2].
OS is a pathological state featuring the over-generation of cellular reactive oxygen and/or nitrogen species (RO/N S) as a result of a dysregulated intracellular metabolism, tissue inflammation or exposure to exogenous stimuli at an extent that exceeds the cellular antioxidant defenses [3]. In fact, OS is considered as a hallmark of carcinogenesis [3].
Regarding CRC, OS on intestinal mucosal cells is proven to damage phospholipid biological membranes via lipid peroxidation by free radicals [4]. Therefore, it causes rupture of the phospholipid bilayer of the cytoplasmic cellular membrane, with the loss of cellular homeostasis. It promotes the destruction of the two lipid bilayers of the endoplasmic reticulum (ER), the dynamic reservoir of Ca2+, leading to disruption of ER-Ca2+ homeostasis, which is implicated in the initiation and propagation of cancer [5]. Also, it causes oxidative damage to the mitochondrial membranes, which disrupts the cell energy metabolism [6].
Another aspect of involvement of OS in CRC pathogenesis is through production of malondialdehyde (MDA, one of the end products of lipid peroxidation), which promotes oxidative DNA damage via formation of adducts with DNA bases; deoxyguanosine and deoxyadenosine. s, thus initiating mutations in tumor suppressor genes and proto-oncogenes [6,7].
Furthermore, free radicals may modify proteins, thus diminishing or enhancing their catalytic activity. Also, free radicals may stimulate cell and tissue lesions by oxidizing carbohydrates [8], and they may exert pro-inflammatory and pro-proliferative roles via oxidizing cholesterol to oxysterols [9]. Interestingly, cancerous cells can maintain certain levels of ROS to promote their proliferation and invasion [4]. Moreover, ROS are key constituents of cancer cells’ survival via the generation of resistance to chemo- and radiotherapies [10]. Accordingly, several studies have indicated that antioxidant compounds were supplemented or tested as adjuvants in cancer therapy to reverse resistance mechanisms, to reduce systemic toxicity during chemo- or radiotherapy [11] and to prevent cancer cell invasion [12].
Chronic inflammation is another principal effector in colorectal carcinogenesis. Inflammation can be induced by different stimuli, including pathogens, toxic compounds and OS-damaged cells. This inflammation enhances the affected tissues to overexpress the inducible cyclooxygenase isoform-2 (COX-2), which is typically absent or rarely expressed in healthy tissues. COX-2 catalyzes the conversion of arachidonic acid (AA) to prostaglandins (PGs) H2, which are further metabolized by prostaglandin synthases to PGD2, E2, I2, F2α and thromboxane A2. Extensive clinical and experimental studies conducted over the last few decades have provided convincing data indicating that high levels of COX-2 and PGE2 are implicated in the pathogenesis of colorectal carcinoma [13,14,15], and that they are associated with poorer survival outcomes [16]. The key roles of the COX-2/PGE2 pathway in CRC pathogenesis include generating an immunosuppressive tumor microenvironment [17], participating in tumor initiation, promoting sustained angiogenesis and thus favoring tumor growth, inducing tumor cell invasion, encouraging migration, reducing apoptotic rate and enhancing chemotherapeutic resistance [18,19]. Therefore, inhibition of COX-2 to reduce PGE2 synthesis is considered an important therapeutic approach in prevention and modulation of CRC progression [20]. Consistent with these findings, a number of meta-analyses and in vitro studies have reported that classical non-steroidal anti-inflammatory drugs (NSAIDs) [21] and selective COX-2 inhibitors [22] provided protection against CRC incidence, reduced mortality and improved chemosensitivity and the efficacy of anticancer drugs when co-administrated [23].
Furthermore, the reprogrammed energy metabolism or Warburg effect [24] is a prominent metabolic characteristic of malignant cells [25]. The common features of this abnormal, less-energy-efficient metabolic pathway include increased glucose uptake, production of two moles of pyruvate and less ATP (2 moles/ glucose molecule) and rapid cytoplasmic fermentation of pyruvate to lactate under catalysis of lactate dehydrogenase A (LDHA). Lactate is then exported to the extracellular environment, which, in the presence of hydrogen ion, generate an acidic tumor microenvironment (TME). Lactic acidosis has been observed in different cancer types and it is linked to tumor aggressiveness [26]. In this regard, several clinical studies have revealed that hyperlactatemia is involved not only in tumor growth but also in the evasion of immune surveillance [27], local invasion, metastasis, chemoresistance and patient survival [28]. Consequently, targeting glycolysis via inhibiting LDHA would suppress hyperlactatemia and prevent its protumorigenic effects, in addition to depriving cancerous cells of nutrients, thereby killing them [29]. Indeed, several studies have shown that inhibition of LDHA activity impedes tumor progression, induces significant oxidative stress and necrosis [30], induces G2/M cell cycle arrest, increases sensitivity to ionizing radiation and 5-Fluorouracil (5-FU) [31,32] and attenuates cell invasion and migration [33].
DM is a multifactorial chronic metabolic disorder affecting millions of people worldwide. DM is categorized into Type 1 (T1DM, caused by the autoimmune destruction of the pancreatic β-cells leading to little, or the absolute lack of, insulin production), Type 2 (T2DM, arises due to complex interactions between genes and some risk factors, mainly obesity, smoking and hypertension, leading to insulin resistance) and gestational (GDM, occurs in pregnant women). T2DM is the most prevalent type, accounting for about 90% of all diabetic cases [34]. Besides its deadly cardiovascular complications, T2DM has also been associated with an increased risk for the development of certain types of cancers [35]. In particular, the positive correlation between CRC and T2DM is attributed to sharing some common risk factors including obesity, alcohol consumption, cigarette smoking and a Western-pattern diet. In addition, genome-wide association studies have identified transcription factor 7-like 2 (TCF7L2), gremlin 1 (GREM1) and tumor protein p53 inducible nuclear protein 1 (TP53INP1), among other genes, as being pleiotropically related to T2DM as well as CRC prognosis and tumorigenesis [36]. Moreover, T2DM-related metabolic changes, such as hyperinsulinemia, hyperglycemia, OS, inflammation induced by adipose tissue dysfunction, impaired immunological surveillance and gastrointestinal motility disorder have been implicated in carcinogenesis [37].
With regard to hyperglycemia, it influences the neoplastic transformations by providing glucose that is essential for the growth and proliferation of cancerous cells under the Warburg state [38]. It leads to the irreversible formation of advanced glycated end-products (AGEs) with proteins, lipids and nucleic acids, as well as their buildup, resulting in the development of OS and chronic inflammations, which are intrinsically linked to induction and development of cancer. Furthermore, elevated expressions of AGEs and their receptors contribute to the invasion and metastasis of CRC [39]. Also, the activations of the polyol, hexosamine and protein kinase C metabolic pathways [40] are other putative mechanisms by which hyperglycemia contributes to the cell proliferation, invasion and tumor progression of colon cancer. In addition, hyperglycemia may negatively affect CRC patients’ quality of life by reducing the chemotherapeutic efficacy and inducing adverse effects [36].
Thus, over the years, researchers have worked on various targets to moderate the onset or worsening of diabetes, from which some antihyperglycemic therapies have been approved by the US FDA and other regulatory agencies, which exhibit different mechanisms of action via targeting single or multiple established molecular pathways involved in the pathogenesis of the disease [41]. Amongst these validated pathways is the breakdown of complex carbohydrates into absorbable monosaccharides, which takes place under the catalysis of alpha-glucosidase (AG) and alpha-amylase (AA). Thus, inhibitors (Is) of these enzymes would manage T2DM by delaying the carbohydrate metabolism and turning down the rate of glucose absorption to reduce the post postprandial blood glucose level (PPBGL). Indeed, carbohydrate mimics [34], including voglibose (natural sugar), miglitol (semisynthetic iminosugar) and 1-deoxynojirimycin (DNJ), all of which inhibit AG, in addition to acarbose (an oligosaccharide), which can also inhibit AA [42], are marketed as antidiabetic drugs. Despite the helpful impacts of these glucose-lowering drugs, morbidity and mortality remain significant in T2DM patients. In addition, these medications have many drawbacks, such as poor compliance with treatment, expense and adverse effects [43,44]. Therefore, there are a lot of synthetic efforts towards the development of new inhibitors, particularly annulated sugars [45], iminosugars [46,47] and novel classes of drugs derived from the structural hybrids of different key molecules and heterocycles [48], with the aim to develop new effective inhibitors that are escorted by better compliance, socioeconomic benefits and safety [49].
Additionally, apoptosis is a well-characterized form of programmed cell death (PCD), which occurs normally for regulating tissue homeostasis and regeneration during development. However, it is also activated as an immune defense mechanism against damaged or stressed cells in response to stimuli such as reactive oxygen species (produced by some chemotherapeutic agents) or DNA damage (from irradiation or anticancer drugs) to remove these potentially harmful cells [50]. If apoptosis in not controlled properly (insufficient apoptosis/extreme cell proliferation), mutations will potentially accumulate in the unwanted cells, which eventually could lead to the induction of cancer. Indeed, resisting apoptosis is considered as one of the hallmarks of cancer [51].
Apoptosis can be triggered by two different pathways: the extrinsic (death receptor) and the intrinsic (mitochondrial) pathways. The extrinsic pathway is initiated via the binding of certain extracellular ligands (members of the tumor necrosis factor super family, TNF) to their cognate cell surface death receptors, resulting in caspase 8 activation. This activation can directly induce apoptosis, or activate effector caspase 3 or Bid, which lead to apoptosis. While the intrinsic apoptotic pathway is initiated via mitochondrial outer membrane permeabilization (MOMP), a process controlled by the members of the B-cell lymphoma (BCL-2) family and p53 proteins [52]. MOMP is considered as the point of no return in this apoptotic cascade, since it leads to the release of the apoptogenic proteins cytochrome c and Smac from the mitochondria into the cell cytoplasm, which activate caspases directly or indirectly, respectively [53,54]. According to the direct mode, cytochrome c interacts with apoptotic protease activating factor-1 (APAF1), thus transforming it into an apoptosome complex, which activates pro-caspase-9 to initiator caspase 9, which in turn activates caspase-3, leading to the activation of the final cascade and, consequently, the fragmentation of the nucleus after its membrane rupture [55]. After the activation of all signaling cascade mediators, the two pathways meet up at the final caspases’ activation (caspases 3, 7), resulting in the cleavage of different integrated cell proteins and the execution of the apoptotic steps [55].
Furthermore, the BCL-2 family of proteins [56], which tightly regulate the intrinsic pathway, is subclassified into three subgroups; the first comprises the pro-apoptotic proteins or apoptosis effectors that positively regulate apoptosis, and they include Bcl-2 antagonist killer 1 (Bak) and Bcl-2-associated X protein (Bax), which can directly execute MOMP when activated.
The second comprises the pro-survival members that negatively regulate apoptosis, and they include B cell lymphoma-2 (Bcl-2) itself, B cell lymphoma extra-large (Bcl-xL), B cell lymphoma-w (Bcl-W), B cell lymphoma-like protein 2 (Bcl-B), myeloid leukemia cell 1 (MCL-1) and Bcl-2-related protein A1/Bcl-2-related isolated from fetal liver-11 (A1/BFL-1).
The third group is the pro-apoptotic BH3-only proteins, which include p53-upregulated modulator of apoptosis (Puma), Bcl-2-interacting mediator of cell death (Bim) and the Bcl-2 Homology 3 interacting death agonist (Bid), which act as activators to Bax and Bak. This group comprises also other proteins, called sensitizers, which do not associate with Bax and Bak, but antagonize the pro-survival members, including the Bcl-2 associated agonist of cell death (Bad), Bcl-2 interacting t killer 1 (Bik), Noxa, BMF and HRK.
In healthy cells, the pro- and anti-apoptotic proteins are held in a fine, delicate balance. Conversely, during cancer development, the malignant cells alter this balance, by overexpressing various anti-apoptotic proteins and/or abnormally reduce the expression levels of pro-apoptotic members, and thus they become unresponsive to stimuli that otherwise trigger apoptosis in sensitive cells, which leads to cellular proliferation and cancer progression, in addition to therapeutic resistance [57]. Therefore, inhibiting the anti-apoptotic [58,59] and activating pro-apoptotic members [60] of the Bcl-2 family have been suggested as plausible selective strategies for cancer therapy and to overcome drug resistance.
Taken together, the complexity of CRC and the manifold and heterogeneous association of this malignancy with OS, inflammation, Warburg effect and hyperglycemia highlight the need for the development of new pharmaceuticals capable of the simultaneous targeting of these risk factors as well as the apoptotic pathways (Figure 1).
Figure 1. Various risk factors and pathways implicated in cancer, which serve as targets for multi-potent agents.
The innovation of multi-targeted therapies represents a promising and prevailing anticancer drug discovery paradigm among medicinal chemists [61]. In line with this principle, quinazolinone scaffolds have proven to be easily accessible pharmacophores, possessing multipotent pharmacological activities both in vitro and in vivo [62]. They have been presented as core structures in inhibitors for; AA [63] I, AG [64] II, and COX-2 [65] III, as well as antioxidant agent [66] IV, and anti-cancer candidates (V, VI and VII), which produce their cytotoxic effects through different molecular mechanisms [63,67,68], as portrayed in Figure 2.
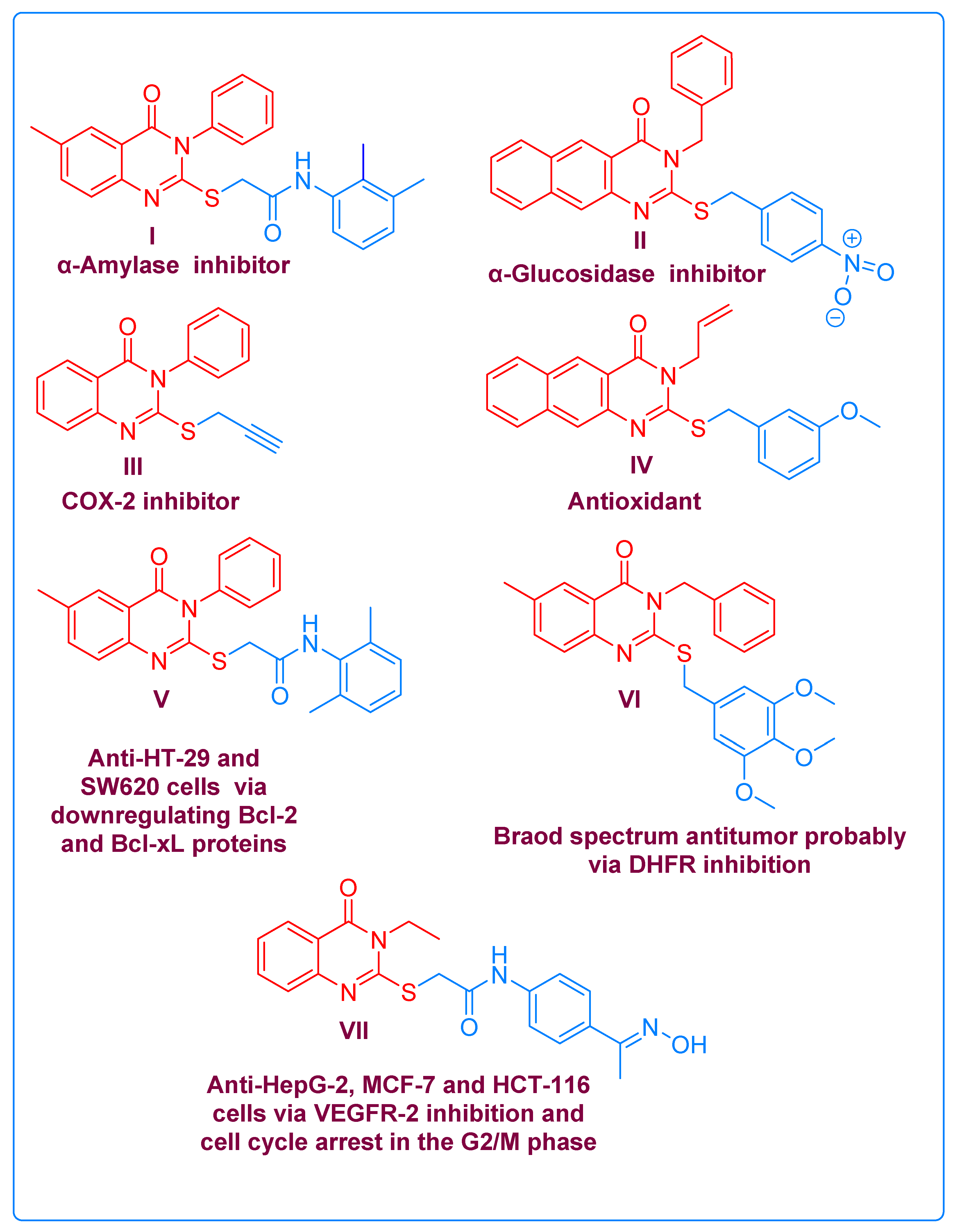
Figure 2. Selected examples of biologically active compounds containing quinazolinone cores.
In light of the above, in this study, we designed a number of new quinazolinone derivatives bearing 4-nitro-phenyl, in addition to fluorine containing 4- or 3-(trifluoromethyl)phenyl moiety at position 3, due to the significant impact of fluorine atoms in enhancing the binding affinity to the targeted protein, as well as improving pharmacokinetic and physicochemical properties including metabolic stability and membrane permeation [69]. Also, various substituents such as Br, Cl and OCH3 were located at the 6 and/or 6,7 positions on the quinazolinone core, to span different electronic, steric and lipophilic characters that would produce different pharmacological responses, which would increase the probability of the discovery of active agents. Subsequently, these compounds were evaluated in vitro as antioxidants as well as AA, AG, COX-2 and LDHA inhibitors. Structure–activity relationships regarding the antioxidant activity were discussed. Additionally, molecular docking simulations were performed to confirm the results of the in vitro enzymatic inhibitory assays and to predict the conformations of the potential inhibitors upon binding to the target proteins, as well as to calculate the binding affinities between them.
Moreover, all compounds were screened for their cytotoxic effects on LoVo and HCT-116 human colon carcinoma cells at 200 µg/mL, and then the IC50 values of the most active cytotoxic candidates were calculated form the concentration–response curves and compared with that of 5-FU. Afterwards, they were evaluated against non-tumoral human umbilical vein endothelial cells (HUVEC) to explore their safety profiles. Furthermore, the capability of the cytotoxic candidates to modulate apoptosis regulators (Bax and BcL2), to activate caspases-8, 9 and 3 and to disturb the cell cycles of LoVo and HCT-116 cells were investigated. Finally, the physicochemical, medicinal chemistry and ADMT properties of all compounds were predicted and discussed in detail for some active compounds as model examples.
2. Results and Discussion
2.1. Chemistry
The target compounds were synthesized as depicted in Scheme 1.
Initially, refluxing mixtures of 5-substituted or 4,5-disubstituted anthranilic acid derivatives 1a-c and 3-substituted or /4-substituted phenyl isothiocyanate derivatives 2a-c in glacial acetic for 10 h [70] yielded the new 6-substituted or 6,7-disubstituted-3-aryl-2-thioxo-2,3-dihydro-1H-quinazolin-4-one derivatives 3a-h, which can potentially also exist in their tautomeric forms 3′a-h due to thiocarbonyl–thiol tautomerization. Subsequently, the arylation of the acidic thiol groups using various aryl halides 4a–d under basic catalysis with potassium carbonate in refluxing dry acetone for 6 h [50] produced eight new 2-(benzylsulfanyl)-3-aryl-3H-quinazolin-4-one derivatives, 5a–h. The structures of the synthesized derivatives were elucidated based on IR, 1H-NMR, 13C-NMR and mass spectrometric analyses.
All the spectroscopic data indicated the predominance of the thiocarbonyl tautomeric form for compounds 3a–h in the solid state (IR) and in the DMSO solution (NMR), which is in agreement with the reported data [71]. Thus, their IR spectra showed stretching absorption bands for the NH groups at a νmax ranging from 3155 to 3245 cm−1. In addition, the 1HNMR spectra demonstrated the NH protons as singlet peaks at δH = 12.91–13.24 ppm, while the 13CNMR spectra exhibited characteristic signals for C= S at a δC/ppm ranging from 174.38 to 176.28.
Contrarily, the IR and 1HNMR spectra of derivatives 5a–h indicted the disappearance of the NH protons, and their 13CNMR spectra confirmed the absence of the peaks corresponding to the C=S groups and the presence of the benzylic methylene groups.
As representative examples, the IR spectrum of compound 3c showed the stretching absorption bands at νmax = 3178 and 1698 cm−1, corresponding to the NH and C=O groups, respectively. The 1H-NMR spectrum of this compound (500 MHz; DMSO-d6) showed a characteristic one-proton singlet peak at δH = 12.91 ppm for the NH proton. Additionally, the four aromatic protons of the 3-trifluoromethylphenyl ring exhibited a one-proton doublet peak (coupling constant J value = 7.5 Hz), a one-proton singlet peak, an apparent triplet peak (J value = 8.05 Hz) and a doublet peak ((J value = 8.5 Hz) at chemical shift values of δH = 7.73, 7.69, 7.66, 7.24 and 7.56 ppm, respectively. The two aromatic protons of the dimethoxyphenyl ring were detected as two singlet peaks at δ = 7.24 and 6.95 ppm.
Lastly, the two methoxy groups exhibited two singlet peaks, each integrating to three protons at δH = 3.83 and 3.77 ppm. With regard to its 13C-NMR spectrum (125 MHz; DMSO-d6), it was characterized by a distinctive peak at δC/ppm = 174.66 attributed to C=S group, in addition to seventeen peaks, as expected, at δC/ppm = 159.34 (C=O), 155.41, 146.58, 140.23, 135.63, 133.62, 130.03, 129.80, 126.38, 125.03, 124.89, 122.87, 108.66, 106.94, 97.98 (6 × CH-aromatic, 6 × Cq-aromatic and CqF3), 56.01 and 55.84 (2 × OCH3). Finally, the MS spectrum of this compound showed a molecular ion peak [M+ + 1] at m/z = 383.17 (27.26%) for C17H13F3N2O3S, and the base peak was detected at m/z = 382.16, which corresponds to [M+].
On the other hand, the IR spectrum (KBr) of compound 5c indicated the disappearance of the stretching absorption band due to NH group of its precursor 3c. The 1H-NMR spectrum (300 MHz, DMSO-d6) endorsed the absence of NH proton and the presence of the characteristic benzylic protons as an AB quartet signal at δH = 4.53 ppm, with a coupling constant of J = 13.5 Hz. In addition, it displayed eight peaks corresponding to ten aromatic protons as follows: a one-proton apparent doublet of the doublet peak (J values = 6.3, 2.1 Hz); an apparent one-proton doublet of the doublet peak (J values = 8.4, 2.4 Hz); a one-proton singlet; a two-proton doublet peak (J = 7.8 Hz); a two-proton multiplet peak; a one-proton triplet peak (J = 7.8 Hz); a one-proton singlet peak and a one-proton singlet peak, which were resonating at δH = 8.52, 8.09, 7.98, 7.93, 7.83–7.77, 7.60, 7.37 and 7.24 ppm, respectively, confirming the installation of the 3-nitrophenyl moiety on the sulfur atoms. The 13C-NMR spectrum (150 MHz; DMSO-d6) revealed the absence of the peak due to the C=S group and the presence a new characteristic CH2-benzylic peak at δC = 35.12 ppm. The mass spectrum of this compound indicated the presence of the molecular ion peaks [M+ + 2] and [M+ + 1] at m/z (%) = 519.10 (6.53) and 518.19 (29.95), respectively, for C24H18F3N3O5S. The base peak, which represents [M+] ion was recorded at m/z = 517.18.
2.2. Biological Evaluation
The synthesized compounds were subjected to multiple in vitro biological evaluations as described in the following sections.
2.3. Molecular Docking Simulations
In order to confirm the results concluded from the in vitro enzymatic inhibitory assays, molecular docking studies were performed. Thus, the relative binding affinities, binding interactions and conformations of the most active candidates within the active sites of their target proteins were identified.
2.4. In Silico Physicochemical, Medicinal and ADMET Predictions
The data for all the synthesized compounds are available in the .
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.3. In Silico Studies
4. Conclusions
Quinazolinone derivatives 3a–h and their S-arylated analogues 5a–h were synthesized and characterized by various spectroscopic techniques. Multi-target biological screening revealed the significance of 3a, 3g and 5a as potent DPPH radical scavengers with lowered IC50 values (mM) of 0.191 ± 0.011, 0.165 ± 0.005 and 0.172 ± 0.004, respectively, as compared with 0.245 ± 0.027 by BHT. Despite this, none of the screened compounds showed an improved inhibitory efficiency to celecoxib (IC50 = 0.136 × 10 −3 ± 0.006 × 10 −3 µM) and sodium oxamate (IC50 = 140.503 ± 7.647 µM) against COX- 2 and LDHA, respectively; quinazolinones 3a (IC50 values = 281.374 ± 10.545 and 273.345 ± 16.087 µM) and 3g (IC50 values = 251.780 ± 22.023 and 242.279 ± 31.298) were recognized as the most active derivatives against these enzymes, respectively. Notably, quinazolinones 3h, 5a and 5h with IC50 values ranging from 12.548 ± 0.542 to 12.882 ± 0.426 µM against AG, and derivatives 3a, 3c, 3f, 3h and 5a–5f as well as 5h, with IC50 values spanning from 186.437 ± 9.700 to 381.335 ± 8.713 µM against AA, exerted superior activity to quercetin (IC50 = 13.126 ± 0.688 and 402.566 ± 10.108, respectively). Therefore, 3h, 5a and 5h can be considered as dual inhibitors for AG and AA.
In addition, the viability assays identified 3a and 3f as the most active antiproliferative agents with IC50 (µM) values of 294.324 ± 8.409 and 383.521 ± 8.989 (LoVo cells), and 298.060 ± 13.247 and 323.596 ± 2.996(HCT-116 cells) as compared with 22.371 ± 0.215(LoVo) and 91.098 ± 2.721(HCT-116) by 5-FU. Interestingly, these compounds did not inhibit the growth of non-tumoral HUVEC at 560.617 and 599.251 µM, with viability percents of 97.000 ± 2.646 and 99.667 ± 0.577, respectively. Quantitative real-time PCR measurements confirmed the capability of 3a and 3f at 10 µg/mL to upregulate the expression levels of Bax and Caspase-3 genes, along with the downregulation of the expression level of the Bcl-2 gene in the cultures of the treated HCT-116 and LoVo cells as compared to the negative control cultures. Moreover, the Western blotting analyses indicated that 3a and 3f were able to induced apoptosis in HCT-116 and LoVo colon cancer cells via pathways triggered by caspases, and predominantly through the intrinsic pathway mediated by caspase-9. Furthermore, the two compounds induced more death for the HCT-116 and LoVo cells via apoptosis than via necrosis; in addition, they induced cell cycle arrest for the LoVo cells in the G2/M phase and for the HCT-116 cells in the G1 phase. Therefore, the observed cytotoxicity of these derivatives could be attributed to the induction of apoptosis and the blocking of cell cycle progression in both cell lines. Molecular docking analyses of the active or potential candidates corroborated the observed inhibitory efficiencies in the in vitro assays against AA, AG, LDAH and COX-2. Lastly, the physicochemical properties, the suitability in medicinal chemistry and the ADMET characteristics were predicted for all the synthesized compounds and explained in detail for some representative active compounds. Collectively, these quinazolinone derivatives may be considered to be suitable candidates for further optimization and exploration against CRC and its risk factors, particularly oxidative stress and DM-type 2.
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Trans. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, G.; Roncucci, L.; Carnevale, G.; Sena, P. Different Roles of Apoptosis and Autophagy in the Development of Human Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 10201. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Z.; Huang, Z.; Nice, E.; Zou, B.; Huang, C. Revisiting cancer hallmarks: Insights from the interplay between oxidative stress and non-coding RNAs. Mol. Biomed. 2020, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Bardelčíková, A.; Šoltys, J.; Mojžiš, J. Oxidative Stress, Inflammation and Colorectal Cancer: An Overview. Antioxidants 2023, 12, 901. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium homeostasis and cancer: Insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4(2), 103–137. [Google Scholar] [CrossRef]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Krejbich, P.; Birringer, M. The Self-Administered Use of Complementary and Alternative Medicine (CAM) Supplements and Antioxidants in Cancer Therapy and the Critical Role of Nrf-2-A Systematic Review. Antioxidants 2022, 11, 2149. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.Y.; Wu, Y.M.; Chang, K.J.; Pan, T.M. Dimerumic acid inhibits SW620 cell invasion by attenuating H2O2-mediated MMP-7 expression via JNK/C-Jun and ERK/C-Fos activation in an AP-1-dependent manner. Int. J. Biol. Sci. 2011, 7, 869–880. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.B.; Li, B.; Zhang, Y.; Zhu, Y.T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Roelofs, H.M.; Te Morsche, R.H.; van Heumen, B.W.; Nagengast, F.M.; Peters, W.H. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014, 14, 9005–9010. [Google Scholar] [CrossRef]
- Wilson, D.J.; DuBois, R.N. Role of Prostaglandin E2 in the Progression of Gastrointestinal Cancer. Cancer Prev. Res. 2022, 15, 355–363. [Google Scholar] [CrossRef]
- Rahman, M.; Selvarajan, K.; Hasan, M.R.; Chan, A.P.; Jin, C.; Kim, J.; Chan, S.K.; Le, N.D.; Kim, Y.B.; Tai, I.T. Inhibition of COX-2 in colon cancer modulates tumor growth and MDR-1 expression to enhance tumor regression in therapy-refractory cancers in vivo. Neoplasia 2012, 14, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Punyawatthananukool, S.; Prasongtanakij, S.; Matsuura, R.; Arima, K.; Nie, H.; Yamamoto, R.; Aoyama, N.; Hamaguchi, H.; Sugahara, S.; et al. PGE2-EP2/EP4 signaling elicits immunosuppression by driving the mregDC-Treg axis in inflammatory tumor microenvironment. Cell Rep. 2022, 39, 110914–110928. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895.e4. [Google Scholar] [CrossRef]
- Sun, Y.; Tang, X.M.; Half, E.; Kuo, M.T.; Sinicrope, F.A. Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res. 2002, 62, 6323–6328. [Google Scholar]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Yu, L.; Xiao, J.; Zhou, X.; Li, X.; Song, P.; Li, X. Aspirin Use and Common Cancer Risk: A Meta-Analysis of Cohort Studies and Randomized Controlled Trials. Front. Oncol. 2021, 11, 690219. [Google Scholar] [CrossRef] [PubMed]
- Arber, N.; Eagle, C.J.; Spicak, J.; Rácz, I.; Dite, P.; Hajer, J.; Zavoral, M.; Lechuga, M.J.; Gerletti, P.; Tang, J.; et al. Celecoxib for the prevention of colorectal adenomatous polyps. N. Engl. J. Med. 2006, 355, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Liu, J.H.; Liu, J.M.; Lin, J.K. Sequence-dependent effect of a cyclooxygenase-2 inhibitor on topoisomerase I inhibitor and 5-fluorouracil-induced cytotoxicity of colon cancer cells. Anti Cancer Drugs 2004, 15, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- El Sayed, S.M. Biochemical Origin of the Warburg Effect in Light of 15 Years of Research Experience: A Novel Evidence-Based View (An Expert Opinion Article). Onco Targets Ther. 2023, 16, 143–155. [Google Scholar] [CrossRef]
- Kliebhan, J.; Besse, A.; Kampa-Schittenhelm, K.; Schittenhelm, M.; Driessen, C. Mutant TP53 driving the Warburg Effect in Mantle Cell lymphoma. Clin. Case Rep. 2022, 10, e6296–e6300. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Daverio, Z.; Balcerczyk, A.; Rautureau, G.J.P.; Panthu, B. How Warburg-Associated Lactic Acidosis Rewires Cancer Cell Energy Metabolism to Resist Glucose Deprivation. Cancers 2023, 15, 1417. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, M.; Kim, H.J.; Jang, S.B.; Bae, S.J.; Lee, I.K.; Ryu, D.; Ha, K.T. Targeting Lactate Dehydrogenase A with Catechin Resensitizes SNU620/5FU Gastric Cancer Cells to 5-Fluorouracil. Int. J. Mol. Sci. 2021, 22, 5406. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, S. Recent Advances in Synthetic α-Glucosidase Inhibitors. Chem. Med. Chem. 2017, 12, 819–829. [Google Scholar] [CrossRef]
- Tomic, D.; Shaw, J.E.; Magliano, D.J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Chang, T.K.; Su, W.C.; Tsai, H.L.; Wang, J.Y. Narrative review of the influence of diabetes mellitus and hyperglycemia on colorectal cancer risk and oncological outcomes. Transl. Oncol. 2021, 14, 101089. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.H.; Li, S.F.; Wei, R.; Jiang, Z. Diabetes and colorectal cancer risk: Clinical and therapeutic implications. J. Diabetes Res. 2022, 2022, 1747326. [Google Scholar] [CrossRef]
- Yeh, C.S.; Wang, J.Y.; Chung, F.Y.; Lee, S.C.; Huang, M.Y.; Kuo, C.W.; Yang, M.J.; Lin, S.R. Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncol. Rep. 2008, 19, 81–91. [Google Scholar] [CrossRef]
- Schröter, D.; Höhn, A. Role of Advanced Glycation End Products in Carcinogenesis and their Therapeutic Implications. Curr. Pharm. Des. 2018, 24, 5245–5251. [Google Scholar] [CrossRef]
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Giglio, R.V.; Ciaccio, M.; Rizzo, M. Diabetes and Colorectal Cancer Risk: A New Look at Molecular Mechanisms and Potential Role of Novel Antidiabetic Agents. Int. J. Mol. Sci. 2021, 22, 12409. [Google Scholar] [CrossRef]
- Dahlén, A.D.; Dashi, G.; Maslov, I.; Attwood, M.M.; Jonsson, J.; Trukhan, V.; Schiöth, H.B. Trends in Antidiabetic Drug Discovery: FDA Approved Drugs, New Drugs in Clinical Trials and Global Sales. Front. Pharmacol. 2022, 12, 807548. [Google Scholar] [CrossRef]
- Ferey-Roux, G.; Perrier, J.; Forest, E.; Marchis-Mouren, G.; Puigserver, A.; Santimone, M. The human pancreatic alpha-amylase isoforms: Isolation, structural studies and kinetics of inhibition by acarbose. Biochim. Biophys. Acta 1998, 1388, 10–20. [Google Scholar] [CrossRef]
- Hedrington, M.S.; Davis, S.N. Considerations when using alpha-glucosidase inhibitors in the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2019, 20, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Pervin, R. Current Antidiabetic Drugs: Review of Their Efficacy and Safety. In Nutritional and Therapeutic Interventions for Diabetes and Metabolic Syndrome, 2nd ed.; Bagchi, D., Nair, S., Eds.; Elsevier Academic Press: Oxford, UK, 2018; Chapter 34, pp. 445–473. [Google Scholar] [CrossRef]
- Rajasekaran, P.; Ande, C.; Vankar, Y.D. Synthesis of (5,6 & 6,6)-oxa-oxa annulated sugars as glycosidase inhibitors from 2-formyl galactal using iodocyclization as a key step. Arkivoc 2022, 2022, 5–23. [Google Scholar] [CrossRef]
- Tseng, P.S.; Ande, C.; Moremen, K.W.; Crich, D. Influence of side chain conformation on the activity of glycosidase inhibitors. Angew. Chem. Int. Ed. Engl. 2023, 62, e202217809. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. (Eds.) Iminosugars: From Synthesis to Therapeutic Applications; John Wiley & Sons Ltd.: Chichester, UK, 2007; pp. 7–123. Available online: http://library.navoiy-uni.uz/files/martin%20o.%20r.%20-%20iminosugars-%20from%20synthesis%20to%20therapeutic%20applications%20(2007)(456s).pdf (accessed on 11 August 2023).
- Singh, A.; Singh, K.; Sharma, A.; Kaur, K.; Kaur, K.; Chadha, R.; Bedi, P.M.S. Recent developments in synthetic α-glucosidase inhibitors: A comprehensive review with structural and molecular insight. J. Mol. Struct. 2023, 1281, 135115. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Zhou, D.; Zheng, G. Targeting anti-apoptotic BCL-2 family proteins for cancer treatment. Future Med. Chem. 2020, 12, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Yangnok, K.; Innajak, S.; Sawasjirakij, R.; Mahabusarakam, W.; Watanapokasin, R. Effects of Artonin E on Cell Growth Inhibition and Apoptosis Induction in Colon Cancer LoVo and HCT116 Cells. Molecules 2022, 27, 2095. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ding, Y.; Ye, N.; Wild, C.; Chen, H.; Zhou, J. Direct Activation of Bax Protein for Cancer Therapy. Med. Res. Rev. 2016, 36, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.G.; Sun, Y.; Sheng, W.B.; Liao, D.F. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef]
- Radwan, A.A.; Alanazi, F.K. Biological activity of quinazolinones. In Quinazolinone and Quinazoline Derivatives; Al-kaf, A.G., Ed.; IntechOpen: London, UK, 2020; Chapter 2; pp. 11–38. Available online: https://www.intechopen.com/chapters/70910 (accessed on 24 February 2023).
- El-Sayed, N.N.E.; Almaneai, N.M.; Ben Bacha, A.; Al-Obeed, O.; Ahmad, R.; Abdulla, M.; Alafeefy, A.M. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3H-quinazolin-4-one derivatives. J. Enzyme Inhib. Med. Chem. 2019, 34, 672–683. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Ahmad, R.; Anouar, E.; Nor Azman, N.I.I.; Marzouk, M.; Abuelizz, H.A. 3-Benzyl(phenethyl)-2-thioxobenzo[g]quinazolines as a new class of potent α-glucosidase inhibitors: Synthesis and molecular docking study. Future Med. Chem. 2018, 10, 1889–1905. [Google Scholar] [CrossRef]
- Moussa, G.; Alaaeddine, R.; Alaeddine, L.M.; Nassra, R.; Belal, A.S.F.; Ismail, A.; El-Yazbi, A.F.; Abdel-Ghany, Y.S.; Hazzaa, A. Novel click modifiable thioquinazolinones as anti-inflammatory agents: Design, synthesis, biological evaluation and docking study. Eur. J. Med. Chem. 2018, 144, 635–650. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Taie, H.A.A.; Bakheit, A.H.; Marzouk, M.; Almehizia, A.A.; Herqash, R.; Abuelizz, H.A. Antioxidant activities and molecular docking of 2-thioxobenzo[g]quinazoline derivatives. Pharmacol. Rep. 2019, 71, 695–700. [Google Scholar] [CrossRef]
- El-Messery, S.M.; Hassan, G.S.; Nagi, M.N.; Habib, E.E.; Al-Rashood, S.T.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of some new methoxylated 2-benzylthio-quinazoline-4(3H)-ones as nonclassical antifolates. Bioorg. Med. Chem. Lett. 2016, 26, 4815–4823. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; El-Gamal, K.M.A.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; Elsohly, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103422. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Hamidian, H.; Tikdari, A.M.; Khabazzadeh, H. Synthesis of New 4(3H)-Quinazolinone Derivatives Using 5(4H)-Oxazolones. Molecules 2006, 11, 377–382. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Ayyad, R.R.; Shawer, T.Z.; Abdel-Aziz, A.A.; El-Azab, A.S. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur. J. Med. Chem. 2016, 112, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Sasada, R.; Chiba, K.; Gotoh, H. Effect of Side Chain Functional Groups on the DPPH Radical Scavenging Activity of Bisabolane-Type Phenols. Antioxidants 2019, 8, 65. [Google Scholar] [CrossRef]
- Kim, E.Y.; Choi, H.J.; Park, M.J.; Jung, Y.S.; Lee, S.O.; Kim, K.J.; Choi, J.H.; Chung, T.W.; Ha, K.T. Myristica fragrans Suppresses Tumor Growth and Metabolism by Inhibiting Lactate Dehydrogenase A. Am. J. Chin. Med. 2016, 44, 1063–1079. [Google Scholar] [CrossRef]
- Lica, J.J.; Wieczór, M.; Grabe, G.J.; Heldt, M.; Jancz, M.; Misiak, M.; Gucwa, K.; Brankiewicz, W.; Maciejewska, N.; Stupak, A.; et al. Effective Drug Concentration and Selectivity Depends on Fraction of Primitive Cells. Int. J. Mol. Sci. 2021, 22, 4931. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.M.; Wu, C.C.; Shyu, R.Y.; Wang, C.H.; Jiang, S.Y. Tazarotene-induced gene 1 inhibits prostaglandin E2-stimulated HCT116 colon cancer cell growth. J. Biomed. Sci. 2011, 18, 88. [Google Scholar] [CrossRef]
- Paul-Samojedny, M.; Kokocińska, D.; Samojedny, A.; Mazurek, U.; Partyka, R.; Lorenz, Z.; Wilczok, T. Expression of cell survival/death genes: Bcl-2 and Bax at the rate of colon cancer prognosis. Biochim. Biophys. Acta 2005, 1741, 25–29. [Google Scholar] [CrossRef]
- Hauseman, Z.J.; Harvey, E.P.; Newman, C.E.; Wales, T.E.; Bucci, J.C.; Mintseris, J.; Schweppe, D.K.; David, L.; Fan, L.; Cohen, D.T.; et al. Homogeneous Oligomers of Pro-apoptotic BAX Reveal Structural Determinants of Mitochondrial Membrane Permeabilization. Mol. Cell 2020, 79, 68–83.e7. [Google Scholar] [CrossRef]
- Wani, A.K.; Akhtar, N.; Mir, T.U.G.; Singh, R.; Jha, P.K.; Mallik, S.K.; Sinha, S.; Tripathi, S.K.; Jain, A.; Jha, A.; et al. Targeting Apoptotic Pathway of Cancer Cells with Phytochemicals and Plant-Based Nanomaterials. Biomolecules 2023, 13, 194. [Google Scholar] [CrossRef]
- Ocker, M.; Höpfner, M. Apoptosis-modulating drugs for improved cancer therapy. Eur. Surg. Res. 2012, 48, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Zorofchian Moghadamtousi, S.; Karimian, H.; Rouhollahi, E.; Paydar, M.; Fadaeinasab, M.; Abdul Kadir, H. Annona muricata leaves induce G₁ cell cycle arrest and apoptosis through mitochondria-mediated pathway in human HCT-116 and HT-29 colon cancer cells. J. Ethnopharmacol. 2014, 156, 277–289. [Google Scholar] [CrossRef]
- Lee, S.T.; Wong, P.F.; Cheah, S.C.; Mustafa, M.R. Alpha-tomatine induces apoptosis and inhibits nuclear factor-kappa B activation on human prostatic adenocarcinoma PC-3 cells. PLoS ONE 2011, 6, e18915. [Google Scholar] [CrossRef]
- Xiang, L.; He, B.; Liu, Q.; Hu, D.; Liao, W.; Li, R.; Peng, X.; Wang, Q.; Zhao, G. Antitumor effects of curcumin on the proliferation, migration and apoptosis of human colorectal carcinoma HCT-116 cells. Oncol. Rep. 2020, 44, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Maurus, R.; Begum, A.; Williams, L.K.; Fredriksen, J.R.; Zhang, R.; Withers, S.G.; Brayer, G.D. Alternative catalytic anions differentially modulate human alpha-amylase activity and specificity. Biochemistry 2008, 47, 3332–3344. [Google Scholar] [CrossRef]
- Hassan, M.Z.; Alsayari, A.; Asiri, Y.I.; Bin Muhsinah, A. 1, 2, 4-Triazole-3-Thiones: Greener, One-Pot, Ionic Liquid Mediated Synthesis and Antifungal Activity. Polycycl. Aromat. Compd. 2023, 43, 167–175. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Chung, T.W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference Compounds. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Duong, V.A.; Maeng, H.J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Boobis, A.; Gundert-Remy, U.; Kremers, P.; Macheras, P.; Pelkonen, O. In silico prediction of ADME and pharmacokinetics. Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci. 2002, 17, 183–193. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting Plasma Protein Binding of Drugs: A New Approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Babalola, S.; Igie, N.; Odeyemi, I. Molecular Docking, Drug-Likeness Analysis, In Silico Pharmacokinetics, and Toxicity Studies of p-Nitrophenyl Hydrazones as Anti-inflammatory Compounds against COX-2, 5-LOX, and H+/K+ ATPase. Pharm. Fronts 2022, 4, e250–e266. [Google Scholar] [CrossRef]
- Bersuder, P.; Hole, M.; Smith, G. Antioxidants from a heated histidine-glucose model system. I: Investigation of the antioxidant role of histidine and isolation of antioxidants by high-performance liquid chromatography. J. Am. Oil Chem. Soc. 1998, 75, 181–187. [Google Scholar] [CrossRef]
- Andrade-Cetto, A.; Becerra-Jiménez, J.; Cárdenas-Vázquez, R. Alfa-glucosidase-inhibiting activity of some Mexican plants used in the treatment of type 2 diabetes. J. Ethnopharmacol. 2008, 116, 27–32. [Google Scholar] [CrossRef]
- Subramanian, R.; Asmawi, M.Z.; Sadikun, A. In vitro alpha-glucosidase and alpha-amylase enzyme inhibitory effects of Andrographis paniculata extract and andrographolide. Acta Biochim. Polym. 2008, 55, 391–398. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
