


1. Introduction
The discovery of new drugs is often facilitated by the inhibition of enzymes. Urease and α-glucosidase play important roles in a variety of medical applications. There are vital enzymes that serve an integral role in various clinical fields [1,2]. There are millions of people worldwide affected by diabetes mellitus (DM), a metabolic disorder caused by high blood sugar levels [3]. A number of complications are associated with diabetes mellitus metabolic disease, such as cardiovascular disorders, reduced wound healing ability, and cancer, as well as severe health problems such as retinopathy, neuropathy, and amputations. Diabetes mellitus can be cured largely through the control of glycemia [4,5,6]. There is a significant amount of α-glucosidase present on the epithelial walls of the small intestines. Carbohydrate metabolism is regulated by it. It is also beneficial to treat diabetes by inhibiting this enzyme, since it reduces the blood’s glucose level [7,8]. Miglitol, acarbose, and voglibose are a few of the medications that can cause flatulence, diarrhoea, and gastrointestinal disease [9,10]. Additionally, it contributes to the conversion of oligosaccharides into simple sugar via hydrolysis in the salivary glands and pancreas. The treatment of diabetes is therefore enhanced by its ability to lower the postprandial blood sugar levels. The high bioavailability of pyrazoline in the organic realm has led to the development of quick, efficient, and environmentally friendly methods for synthesizing it [11].
Urease belongs to the super family of phosphodiesterase and amidohydrolase, which initiate the degradation of urea into ammonia and carbon dioxide. Eukaryotes and prokaryotes both produce urease enzymes. In response to the hyperactivity of the urease enzyme, excessive amounts of ammonia are released into the stomach, causing alkalinity and helping to improve mucosal permeability [12,13,14,15]. Even though humans lack urease enzymes, they synthesize urea as an end product of protein metabolism and eliminate it through urine [16,17,18]. Urease enzymes facilitate monitoring and regulating the nitrogen metabolism in cattle and many other animals. In addition to causing bacterial infections, high levels of these enzymes can cause various pathogenic conditions. In humans, low stomach pH eases the growth of Helicobacter pylori (HP). In the long run, it causes ulcers in the stomach and intestines, and ultimately may lead to cancer [19,20,21,22,23].
Individuals who concurrently have diabetes mellitus and peptic ulcers are advised to employ a variety of medical therapies. The coexistence of a peptic ulcer can potentially amplify health concerns in patients diagnosed with diabetes due to the commonly seen phenomenon of delayed wound healing. A proposed approach has been formulated to address the management of individuals who have both diabetes mellitus and peptic ulcers. This technique aims to simultaneously target the activities of α-glucosidase and urease. This technique holds significant potential as a therapeutic alternative for people with DM and peptic ulcers [24,25].
Pyrazole-based compounds have garnered significant attention owing to their diverse antineoplastic properties, including chemotherapy. The presence of a pyrazole core has been recognized as a significant contributor to the augmented cytotoxicity of chalcones [26].
In recent times, there has been a notable surge in interest surrounding pyrazoline as a promising heterocyclic framework within the realm of medicine. This heightened attention can be attributed to the widespread use of pyrazoline in the development of therapeutic medications. The compound has a significant role in various fields, such as synthetic and medicinal chemistry, serving as a crucial foundational component for the production of pharmaceuticals. Heterocyclic compounds have been found to have an affinity towards a diverse range of biotargets across several fields [27,28,29]. Both agribusiness and pharmacological research have derived advantages from the utilization of these scaffolds [30].
There have been numerous reports of thiazole and pyrazoline scaffolds exhibiting a broad range of chemical reactivity and pharmacological activities for decades [31,32]. Numerous studies have demonstrated that these five-membered heterocyclic compounds delocalize free radicals and inhibit bacteria by producing stable DPPH fragments [33]. Thiazole-based pyrazoline scaffolds also exhibit anti-inflammatory [34], antidiabetic [35], Alzheimer’s disease [36], anti-HIV [37], antiviral [38], anticonvulsant, antitumor activities [39], and antimicrobial properties [40]. The pyrazoline skeleton has been extensively studied using a variety of structural manipulations and it has been concluded that the steric and electronic properties of substitutes at the N-1, C-3, and C-5 positions, as well as their chirality, have significant effects on their biological activity [41]. A pyrazoline’s N-N bonds are said to play an important role in its pharmacological properties [42].
Pyrazoline structures have been found to possess noteworthy pharmacological properties, encompassing anti-inflammatory, antioxidant, analgesic, antifungal, antipyretic, antiangiogenic, antibacterial, antitumor, and antiviral activities [43,44]. Our research group is presently involved in the development of novel and more effective synthetic routes for small compounds. This endeavour aims to enable the investigation of their potential biological activities, specifically their inhibitory effects on enzymes such as α-glucosidase and urease, which have been observed to be significant.
As pyrazoline structural patterns play an important role in medicinal chemistry, we present our findings concerning the synthesis, characterization, and assessment of novel pyrazoline derivatives. As dual inhibitors, these derivatives inhibit both α-glucosidase and urease enzymes. A variety of pyrazoline derivatives [45,46] containing different substitution arrangements were evaluated for their ability to inhibit both α-glucosidase and urease enzymes. Among heterocyclic compounds, these analogues have shown a considerable potency, providing a greater pool of potential of α-glucosidase and urease inhibitors than previously known (Figure 1).
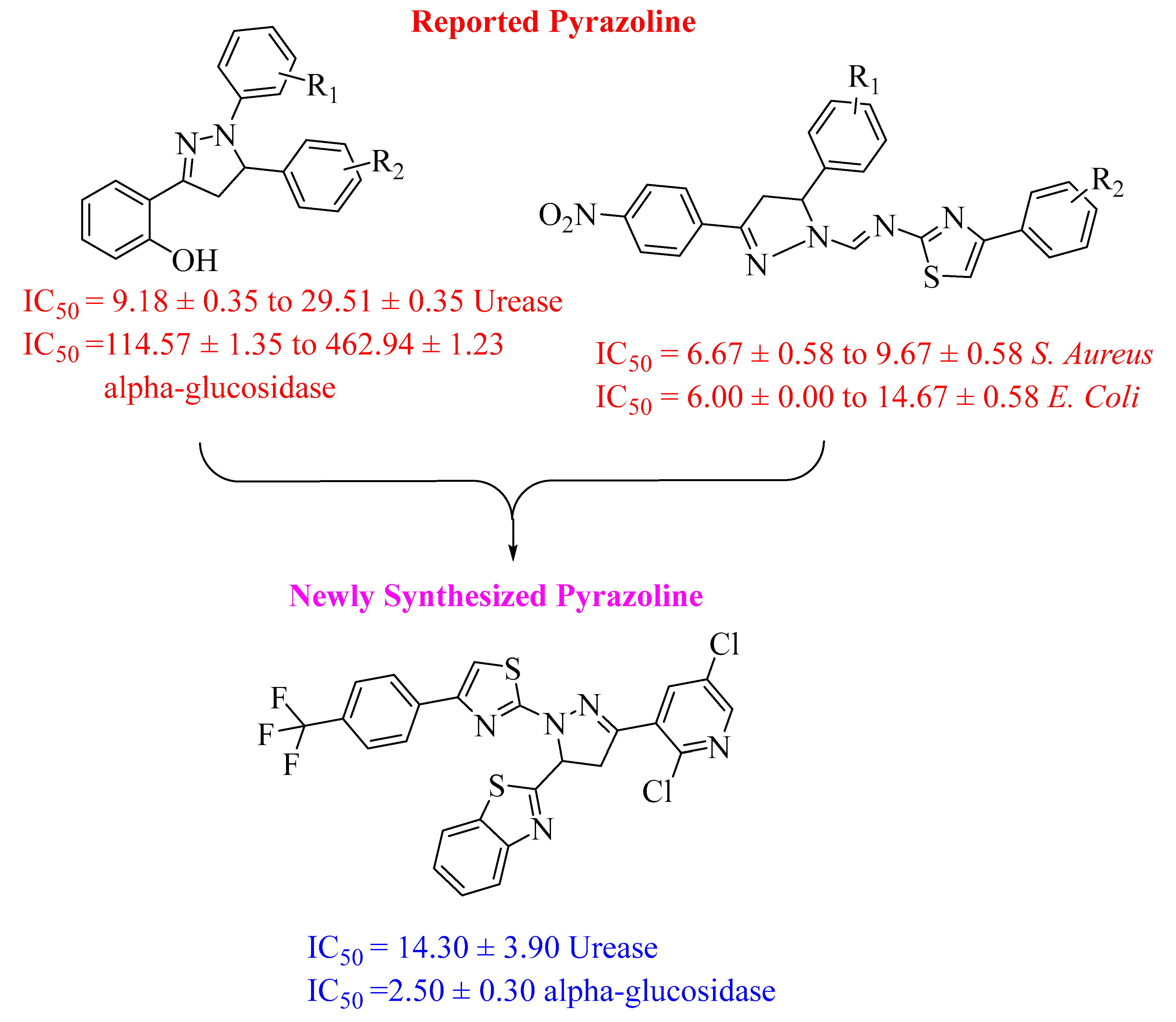
Figure 1. Rational study of the synthesized pyrazoline-based thiazole analogues.
2. Results and Discussion
2.1. Chemistry
In order to synthesize pyrazoline-based thiazoles, a multi-step reaction procedure was involved. The desired pyrazoline-based thiazoles derivatives (1−17) were synthesized using the method outlined in Scheme 1. In the first step, 1-(2,5-dichloropyridin-3-yl)ethan-1-one (I) was reacted with benzo[d]thiazole-2-carbaldehyde (II) to produce a substrate chalcone (III) [47]. This reaction was carried out in the presence of a 50% sodium hydroxide solution in absolute ethanol. It was refluxed for 8–10 h continuously with constant stirring in an ice bath. The substrate (III) was neutralized with dilute hydrochloric acid and washed with pet-ether. The substrate (III) endured cyclization by reacting with thiosemicarbazide that was stirred in acetic acid, and heated under reflux conditions for 5–7 h, enabling the formation of an intermediate pyrazoline (IV). Finally, the substrate (IV) was reacted with differently substituted phenacyl bromide in the presence of triethyl amine (Et3N) in solvent absolute ethanol, affording the targeted pyrazoline-based thiazole derivatives (V) in an appropriate yield (Scheme 1).
2.2. Biological Activities
2.3. Molecular Docking Studies
In this study, docking studies enable us to identify robust binding interactions between specific ligands and enzyme active sites. The investigation involved various substitutions in pyrazoline-based thiazole scaffolds (1–17). These compounds were shown to be effective against the targeted proteins through molecular docking analyses. As a result of using a variety of software tools, we were able to visualize the interaction between ligands and amino acids, determining their distances and interaction types, such as C-H bonds, pi-R interactions, pi-S interactions, hydrogen bonds, pi-pi T-shaped interactions, pi-stacking, and pi-sigma interactions, which are shown below in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11 and Figure 12 and . Our research team synthesized various heterocyclic compounds with a variety of substitutions and evaluated their potential biological activity using diverse molecular docking software [48,49,50,51,52,53].
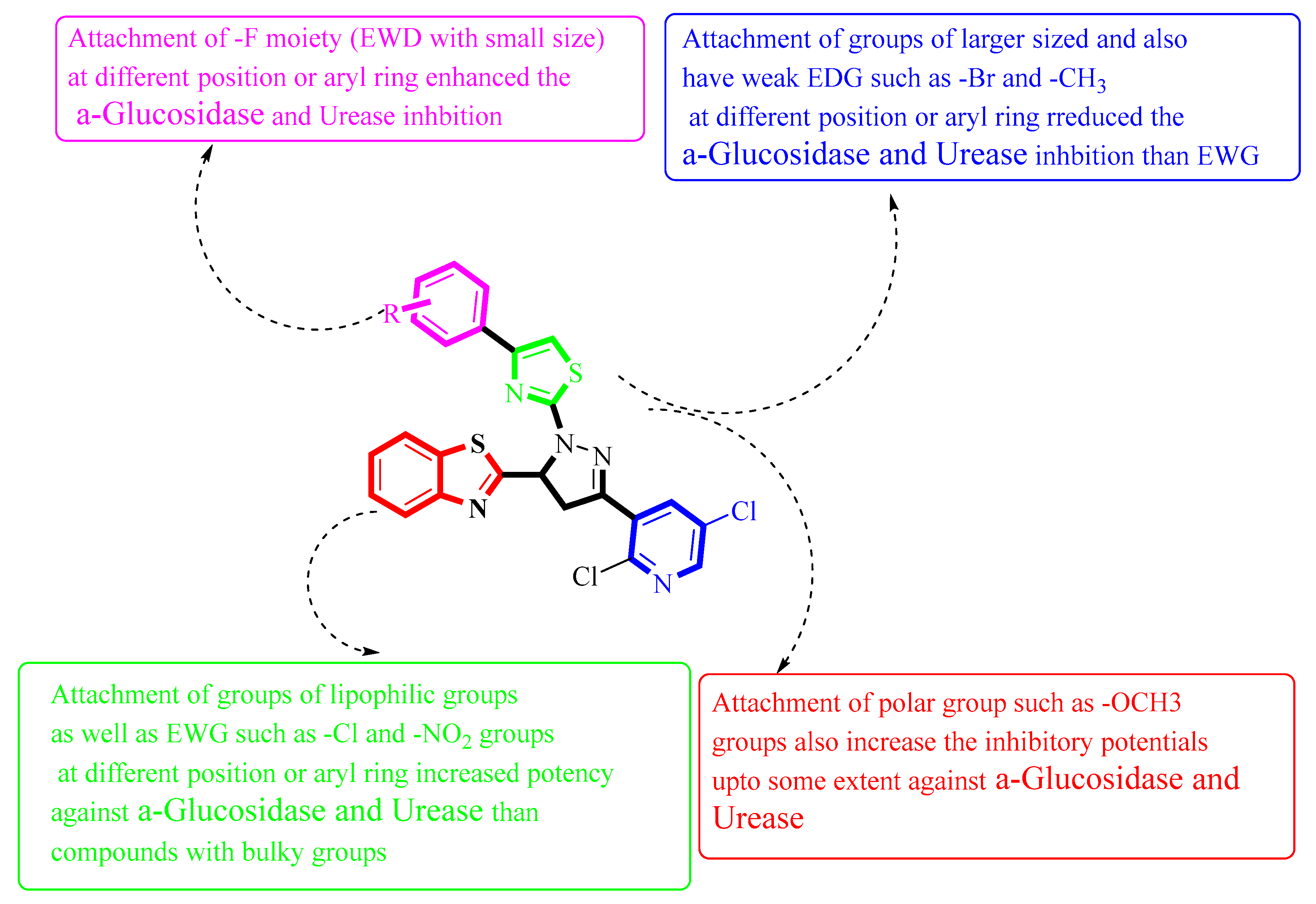
Figure 2. Summary of SAR studies of pyrazoline–thiazole hybrids analogues against α–glucosidase and urease enzymes.
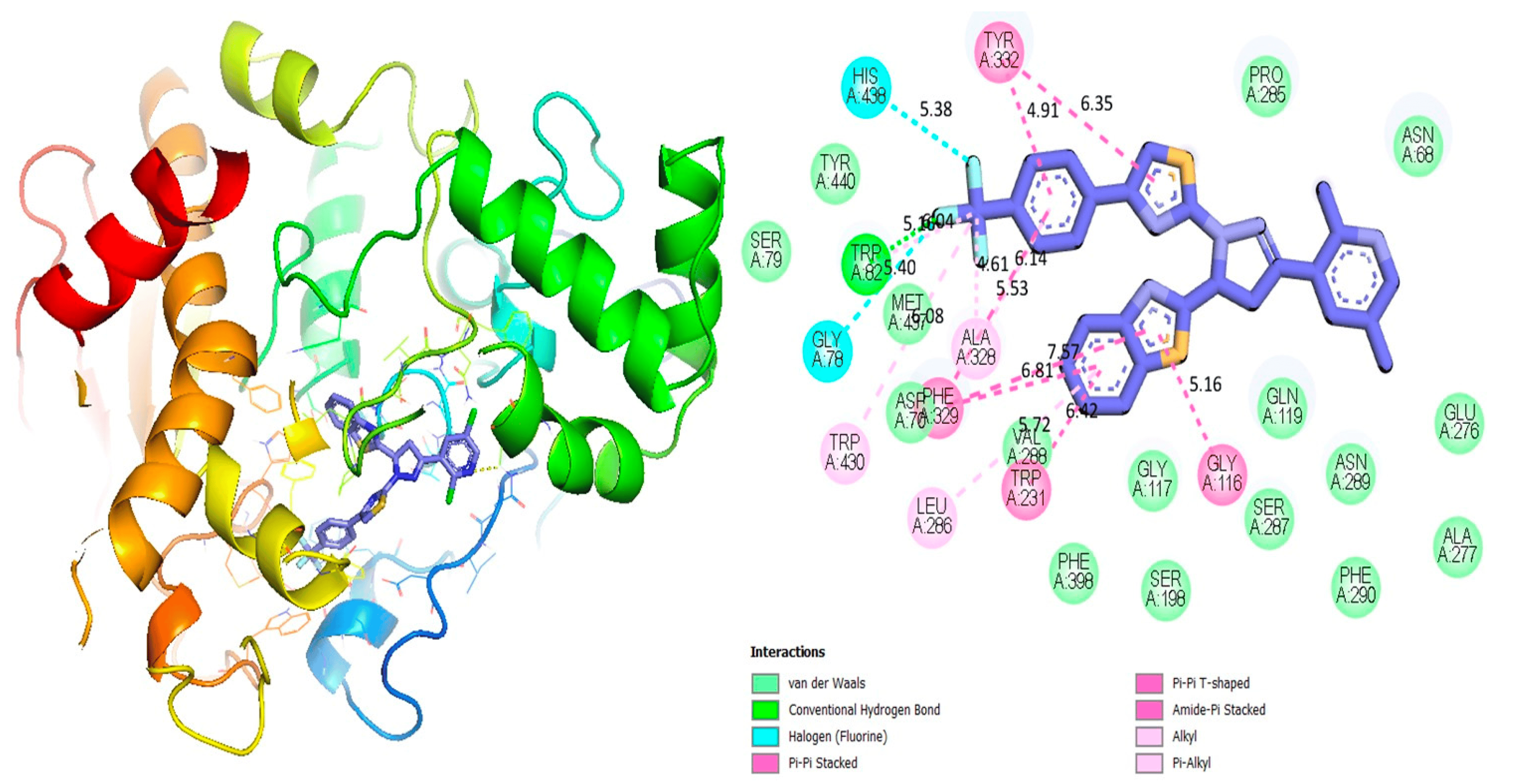
Figure 3. Protein–ligand interaction 2D and 3D profile of an active analogue 6 against α-glucosidase.
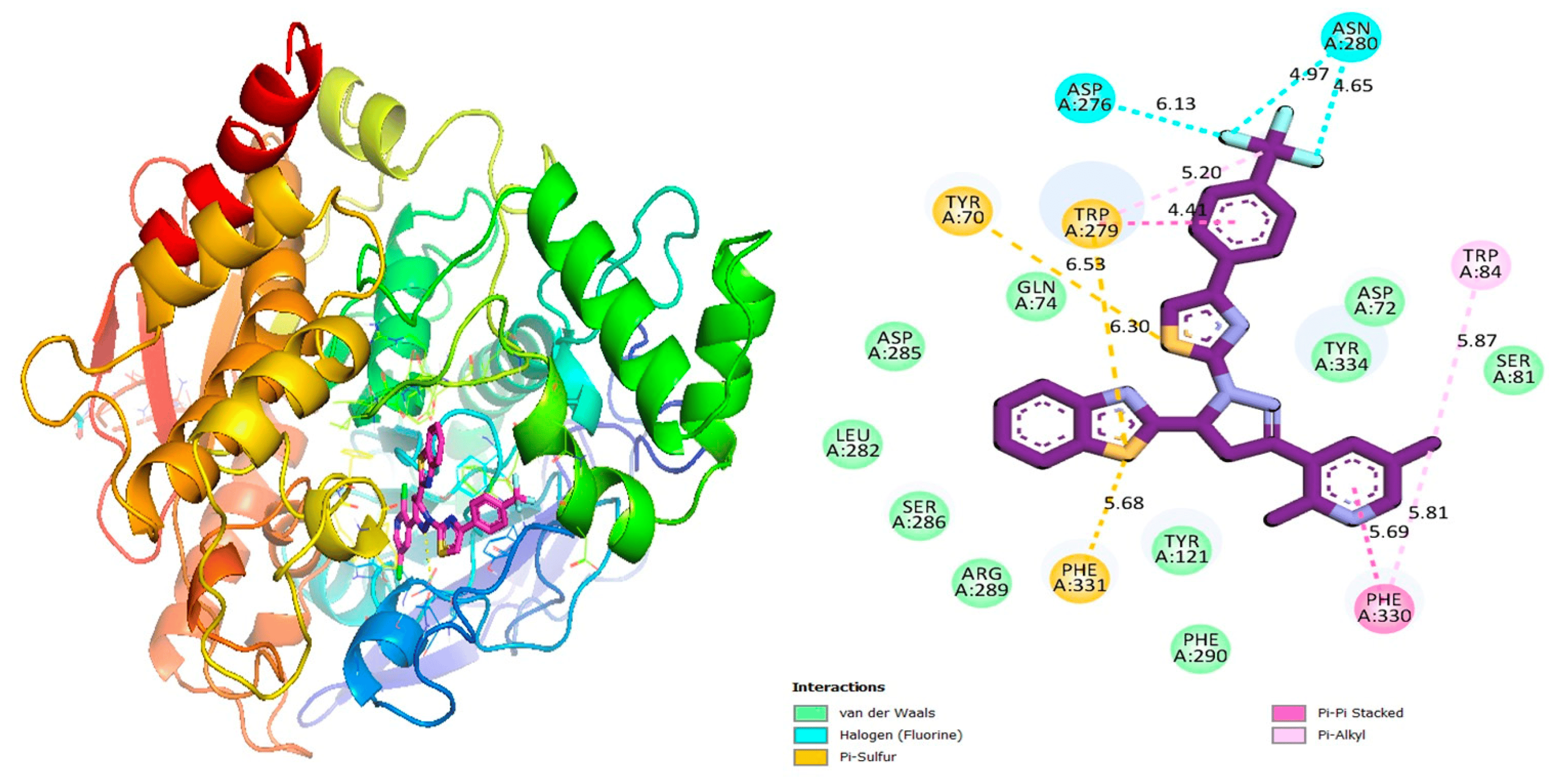
Figure 4. Protein–ligand interaction 2D and 3D profile of an active analogue 6 against urease.
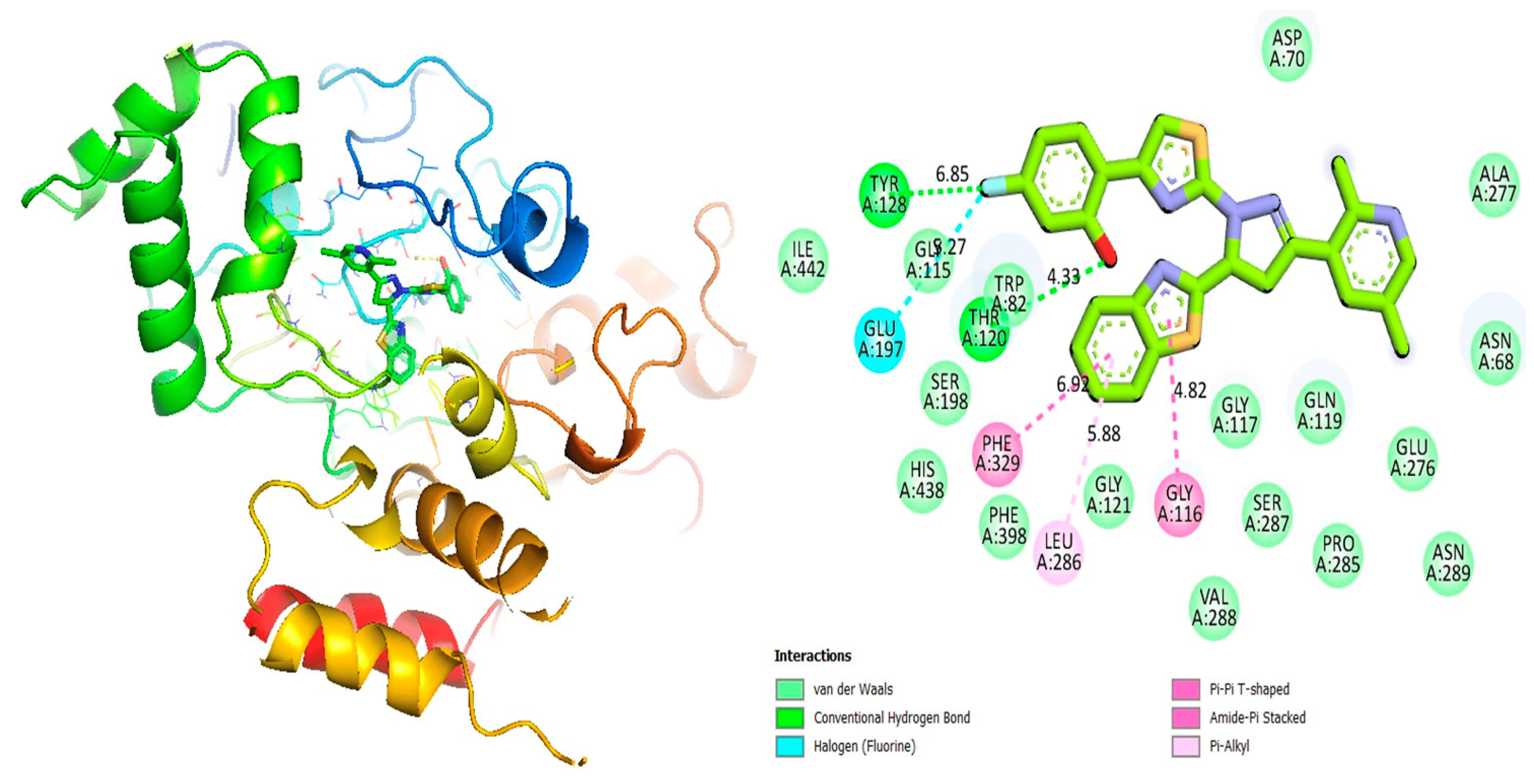
Figure 5. Protein–ligand interaction 2D and 3D profile of an active analogue 7 against α-glucosidase.
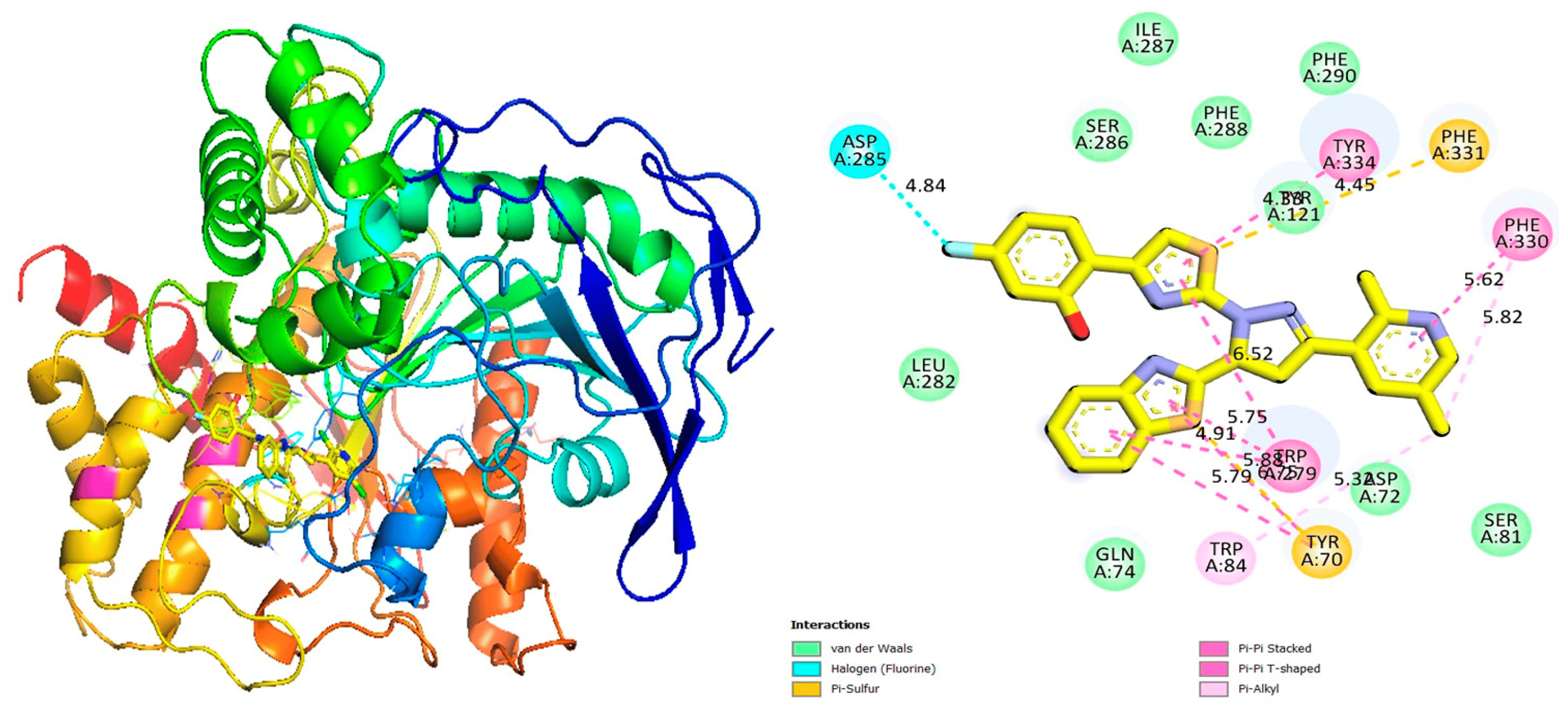
Figure 6. Protein–ligand interaction 2D and 3D profile of an active analogue 7 against urease.
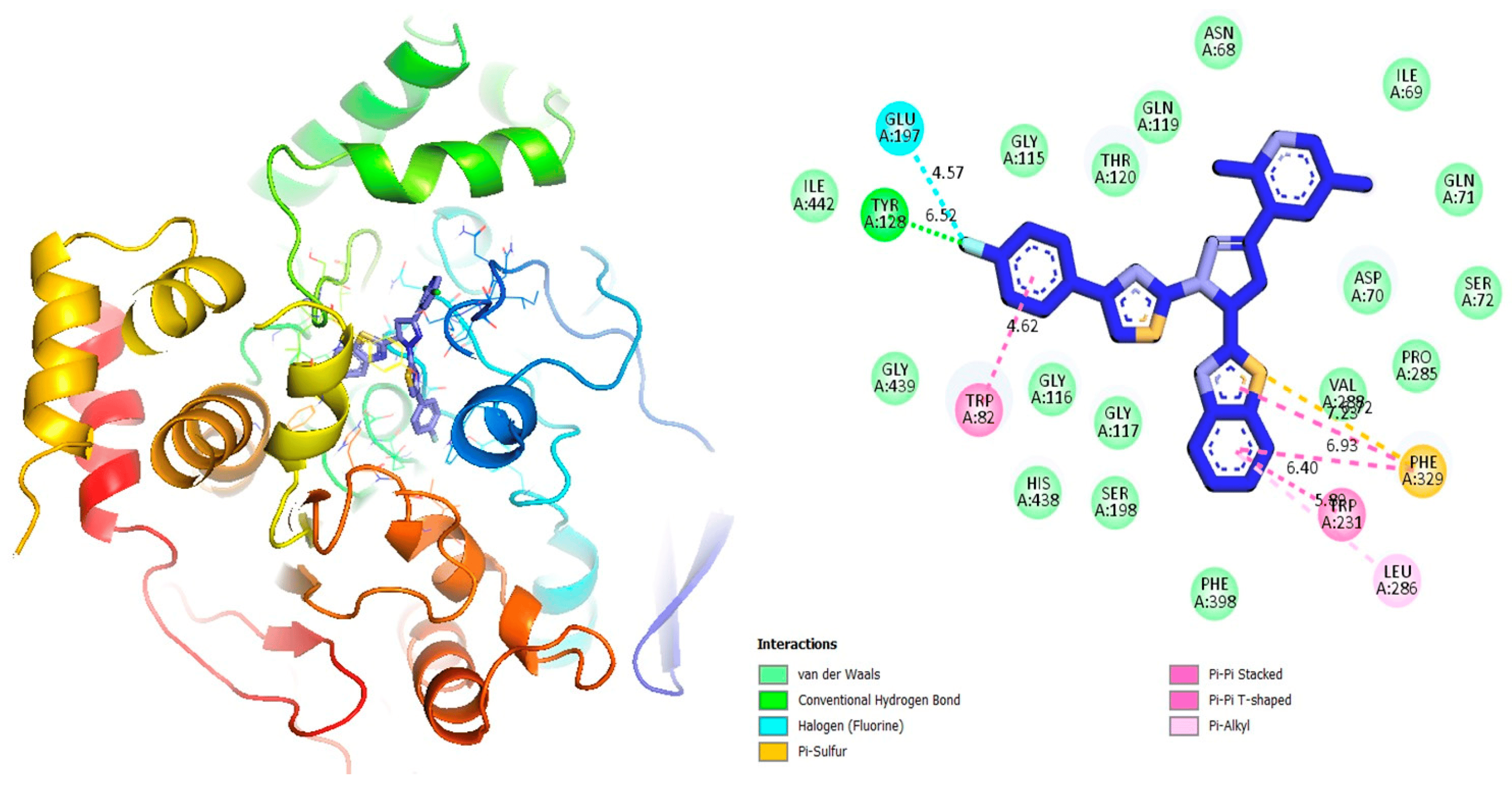
Figure 7. Protein–ligand interaction 2D and 3D profile of an active analogue 12 against α-glucosidase.
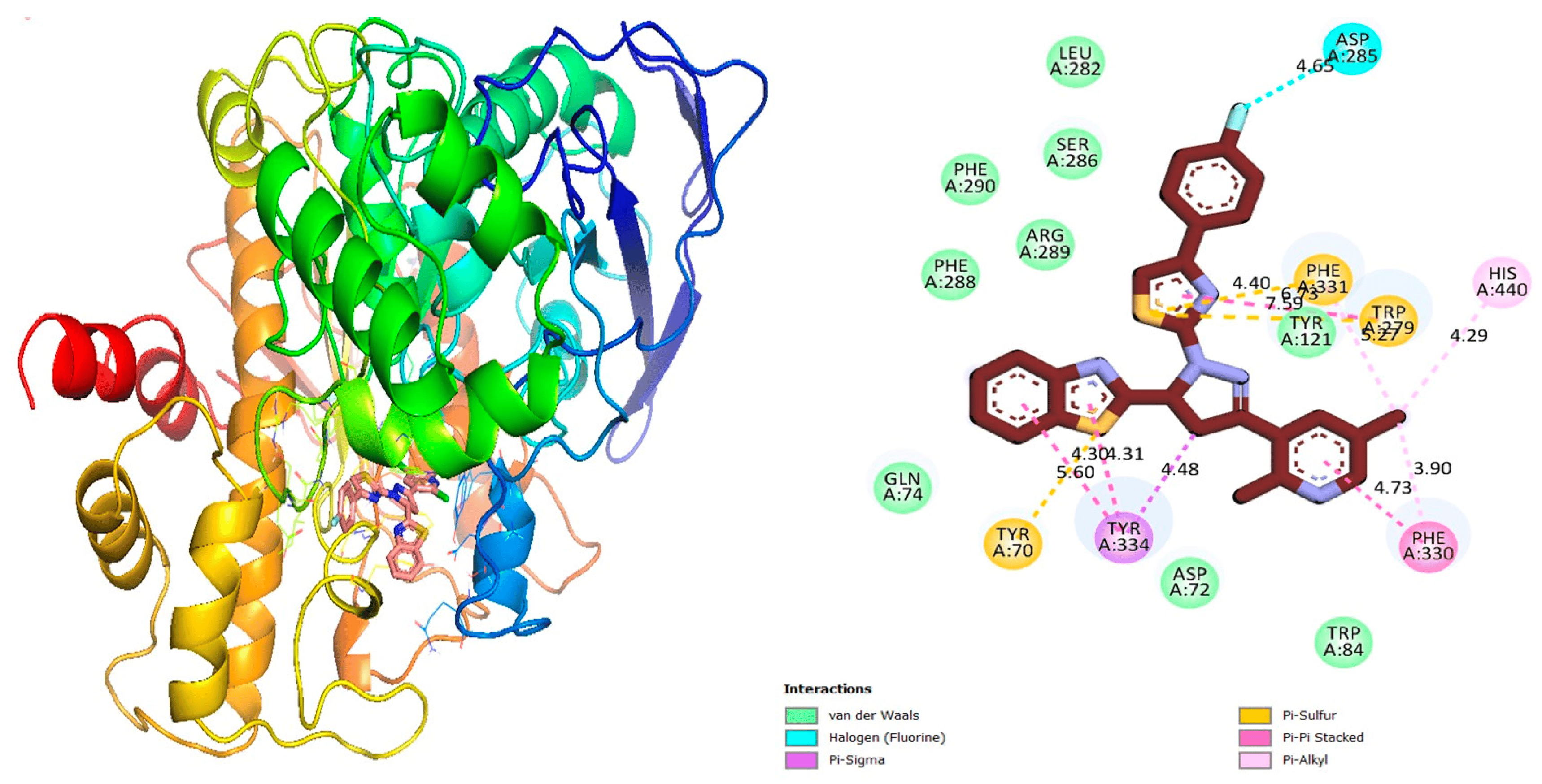
Figure 8. Protein–ligand interaction 2D and 3D profile of an active analogue 12 urease.
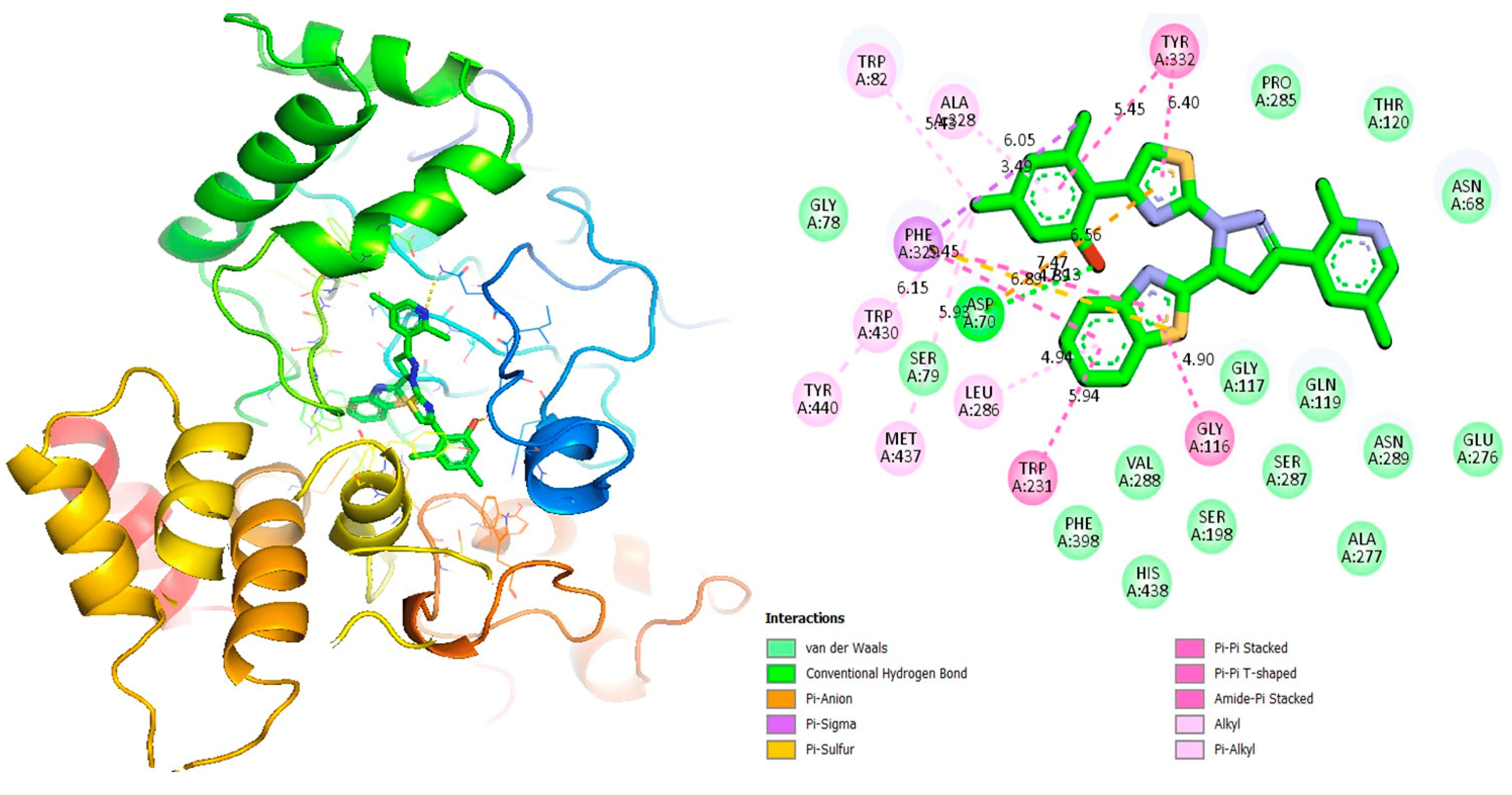
Figure 9. Protein–ligand interaction 2D and 3D profile of an active analogue 14 against α-glucosidase.
3. Materials and Methods
3.1. General Information
This study used chemicals and solvents that were obtained from reputable suppliers, namely Merck and Sigma-Aldrich, without any further purification. An electrothermal digital device was used to determine the melting points. A Bio-Rad spectrophotometer was used to acquire the infrared spectra. Bruker spectrometers with proton frequencies of 600 MHz and carbon frequencies of 126 MHz were used to acquire the NMR spectra. Nuclear magnetic resonance (NMR) chemical shift values were first established using the δ (parts per million) scale. Thin-layer chromatography (TLC) was employed as a means of assessing and overseeing the advancement and culmination of the reaction. The spots on the TLC plate were visualized by exposing it to 254 nm ultraviolet light (UV). In addition to DMSO-d6, a reference compound was used to calculate chemical shift values. Moreover, the coupling constants are expressed in hertz (Hz). We acquired mass spectra using the mass spectrometers MAT 113D, JEOL JMS-600H, and MAT 312 (high-resolution electron ionization mass spectrometry (HR-EI-MS).
3.2. General Procedure for the Synthesis of Chalcone
3.3. Spectral Analysis
3.4. Molecular Modelling Assay
In this study, proteins and ligands were examined using the software AutoDock 4.2.0. Models were obtained from the RCSB Protein Data Bank (PDB ID: 5NN6), which contained the 2-(2-)-2-(hydroxymethyl)piperidine-3,4,5-triol). AHA and MIG were created in the binding spaces of urease and α-glucosidase using Discovery Studio 4.0. AHAs and MIGs were then refined and hydrogens were added to the side-chains [48,49,50,51,52].
3.5. α-Glucosidase Inhibitory Assay
A procedure based on the previously reported procedure with minor modifications was used to determine the inhibitory capacity of the newly designed derivatives of benzothiazole-derived pyrazoline-based thiazole (1–17). For 20 min at 37 °C, 250 mL of 1.0 U/mL carbose was incubated with 500 mL of 1.0 U/mL glucose in 100 mM of phosphate buffer (pH 6.8) for 20 min. The reaction mixture was then incubated for 1 h at 37 °C with 250 mL of 4-nitrophenyl-b-D-glucopyranoside solution and 250 mL of 1% starch dissolved in 100 mM of phosphate buffer (pH 6.8), respectively. Following this, the reaction mixture was heated for 10 min with 1.0 mL of 3,5-dinitrosalicylic acid colouring reagent. The absorbance of the final reaction mixture was determined for α-glucosidase at 405 nm. To correct the absorbance, a blank was prepared. The positive control used was acarbose solution. A percentage of the inhibitory activity was calculated using a control sample without the inhibitors [53].
3.6. Urease Inhibitory Assay
In this study, newly synthesized pyrazoline-based thiazole derivatives (1–17) were evaluated for their ability to inhibit urease activity. During the experiment, 55 L of buffer solution containing 100 mM of urea was combined with 25 L of enzyme solution to prepare reaction mixtures. A volume of 5 L containing 1 mM concentrations of recently produced compounds was then added to these mixtures. After 15 min of incubation at 30 °C, the reactions were allowed to occur in 96-well plates. The detection of ammonia generation by urease was carried out by using the Indophenol technique. A phenol reagent containing 1% w/v phenol and 0.005% w/v sodium nitroprusside was added to each well along with 70 lL of an alkali reagent containing 0.5% w/v NaOH and 0.1% active chloride NaOCl. Utilizing a microplate reader manufactured by Molecular Device in the United States, the increased absorbance was measured at 630 nm after another 50 min. In triplicate, the aforementioned procedures were performed using a constant end volume of 200 mL. The final data, representing the rate at which the absorbance changed per minute, were analysed with the SoftMax Pro program by Molecular Device, New York, NY, USA. In alkaline conditions with a pH of 8.2, the experiments were conducted using 0.01 M K2HPO4·3H2O, 1 mM EDTA, and 0.01 M LiCl. A conventional inhibitor of urease, thiourea, was used in this study to determine the percentage of inhibition [54].
4. Conclusions
A series of newly benzothiazole-derived pyrazoline-based thiazole derivatives (1–17) were designed and synthesized. The synthesized benzothiazole-derived pyrazoline-based thiazole compounds were characterized through different spectroscopic techniques such as 1H-NMR, 13C-NMR, and HRMS. After that, their inhibitory activity against α-glucosidase and urease enzymes was evaluated. The compounds (1–17) exhibited remarkable inhibitory potential against targeted α-glucosidase and urease enzymes under the positive control of acarbose and thiourea as standard drugs, respectively. Among the synthesized series, the compounds 6, 7, 12, and 14 demonstrated were found to be significantly active, as seen by their very low IC50 values, indicating their notable inhibitory activity. Furthermore, the structure–activity relationship (SAR) study was carried out based on various functional groups linked to the aryl group, the nature of the substituent, and positions of the substituent, which play a significant role in determining the effectiveness of these compounds as inhibitors of α-glucosidase and urease enzymes. In addition, to correlate the in vitro study well with the in silico study, a molecular docking study was conducted for the most active compounds and the result obtained corroborated that these active compounds established several key interactions with the active sites of targeted enzymes.
References
- Arshad, T.; Khan, K.M.; Rasool, N.; Salar, U.; Hussain, S.; Asghar, H.; Ashraf, M.; Wadood, A.; Riaz, M.; Perveen, S.; et al. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of α-glucosidase and urease enzymes. Bioorganic Chem. 2017, 72, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Taha, M.; Ullah, H.; Wadood, A.; Selvaraj, M.; Rab, A.; Sajid, M.; Shah, S.A.A.; Uddin, N.; Gollapalli, M. Synthesis of new arylhydrazide bearing Schiff bases/thiazolidinone: α-Amylase, urease activities and their molecular docking studies. Bioorganic Chem. 2019, 91, 103112. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, F.M.; Daneshfar, M.; Daneshfar, Z.; Iraji, A.; Samandari-Najafabad, A.; Faramarzi, M.A.; Mahdavi, M. Synthesis and characterization of 1-amidino-O-alkylureas metal complexes as α-glucosidase Inhibitors: Structure-activity relationship, molecular docking, and kinetic studies. J. Mol. Struct. 2022, 1250, 131726. [Google Scholar] [CrossRef]
- Azimi, F.; Azizian, H.; Najafi, M.; Hassanzadeh, F.; Sadeghi-Aliabadi, H.; Ghasemi, J.B.; Faramarzi, M.A.; Mojtabavi, S.; Larijani, B.; Saghaei, L.; et al. Design and synthesis of novel quinazolinone-pyrazole derivatives as potential α-glucosidase inhibitors: Structure-activity relationship, molecular modeling and kinetic study. Bioorganic Chem. 2021, 114, 105127. [Google Scholar] [CrossRef]
- Xie, H.X.; Zhang, J.; Li, Y.; Zhang, J.H.; Liu, S.K.; Zhang, J.; Zheng, H.; Hao, G.Z.; Zhu, K.K.; Jiang, C.S. Novel tetrahydrobenzo [b] thiophen-2-yl) urea derivatives as novel α-glucosidase inhibitors: Synthesis, kinetics study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorganic Chem. 2021, 115, 105236. [Google Scholar] [CrossRef]
- Gani, R.S.; Kudva, A.K.; Timanagouda, K.; Mujawar, S.B.H.; Joshi, S.D.; Raghu, S.V. Synthesis of novel 5-(2,5-bis (2,2,2-trifluoroethoxy) phenyl)-1,3,4-oxadiazole-2-thiol derivatives as potential glucosidase inhibitors. Bioorganic Chem. 2021, 114, 105046. [Google Scholar] [CrossRef]
- Raghu, C.; Arjun, H.A.; Anantharaman, P. In vitro and in silico inhibition properties of fucoidan against α-amylase and α-D-glucosidase with relevance to type 2 diabetes mellitus. Carbohydr. Polym. 2019, 209, 350–355. [Google Scholar]
- Alqahtani, A.S.; Hidayathulla, S.; Rehman, M.T.; ElGamal, A.A.; Al-Massarani, S.; Razmovski-Naumovski, V.; Alqahtani, M.S.; El Dib, R.A.; AlAjmi, M.F. Alpha-amylase and alpha-glucosidase enzyme inhibition and antioxidant potential of 3-oxolupenal and katononic acid isolated from Nuxia oppositifolia. Biomolecules 2019, 10, 61. [Google Scholar] [CrossRef]
- Goff, H.D.; Repin, N.; Fabek, H.; El Khoury, D.; Gidley, M.J. Dietary fibre for glycaemia control: Towards a mechanistic understanding. Bioact. Carbohydr. Diet. Fibre 2018, 14, 39–53. [Google Scholar] [CrossRef]
- Xiao, J.B.; Hogger, P. Dietary polyphenols and type 2 diabetes: Current insights and future perspectives. Curr. Med. Chem. 2015, 22, 23–38. [Google Scholar] [CrossRef]
- Kumar, L.; Lal, K.; Yadav, P.; Kumar, A.; Paul, A.K. Synthesis, characterization, α-glucosidase inhibition and molecular modeling studies of some pyrazoline-1H-1, 2, 3-triazole hybrids. J. Mol. Struct. 2020, 1216, 128253. [Google Scholar] [CrossRef]
- Ahmed, M.; Imran, M.; Muddassar, M.; Hussain, R.; Khan, M.U.; Ahmad, S.; Mehboob, M.Y.; Ashfaq, S. Benzenesulfonohydrazides inhibiting urease: Design, synthesis, their in vitro and in silico studies. J. Mol. Struct. 2020, 1220, 128740. [Google Scholar] [CrossRef]
- Shi, W.K.; Deng, R.C.; Wang, P.F.; Yue, Q.Q.; Liu, Q.; Ding, K.L.; Yang, M.H.; Zhang, H.Y.; Gong, S.H.; Deng, M.; et al. 3-Arylpropionylhydroxamic acid derivatives as Helicobacter pylori urease inhibitors: Synthesis, molecular docking and biological evaluation. Bioorganic Med. Chem. 2016, 24, 4519–4527. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Khan, K.M.; Zehra, S.T.; Ahmed, R.; Shafiq, Z.; Bakht, S.M.; Yaqub, M.; Hussain, M.; de León, A.D.L.V.; Furtmann, N.; et al. Synthesis, biological evaluation and molecular docking of N-phenyl thiosemicarbazones as urease inhibitors. Bioorganic Chem. 2015, 61, 51–57. [Google Scholar] [CrossRef]
- Holter, H.; Linderstrøm-Lang, K. Micromethods and their application in the study of enzyme distribution in tissues and cells. Physiol. Rev. 1951, 31, 432–448. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Khan, A.; Shah, S.A.A.; Anwar, A.; Halim, S.A.; Fatmi, M.Q.; Imran, S.; Rahim, F.; Khan, K.M. Synthesis of novel derivatives of oxindole, their urease inhibition and molecular docking studies. Bioorganic Med. Chem. Lett. 2015, 25, 3285–3289. [Google Scholar] [CrossRef]
- Shehzad, M.T.; Khan, A.; Islam, M.; Halim, S.A.; Khiat, M.; Anwar, M.U.; Hussain, J.; Hameed, A.; Pasha, A.R.; Khan, F.A.; et al. Synthesis, characterization and molecular docking of some novel hydrazonothiazolines as urease inhibitors. Bioorganic Chem. 2020, 94, 103404. [Google Scholar] [CrossRef]
- Shehzad, M.T.; Khan, A.; Islam, M.; Hameed, A.; Khiat, M.; Halim, S.A.; Anwar, M.U.; Shah, S.R.; Hussain, J.; Csuk, R.; et al. Synthesis and urease inhibitory activity of 1, 4-benzodioxane-based thiosemicarbazones: Biochemical and computational approach. J. Mol. Struct. 2020, 1209, 127922. [Google Scholar] [CrossRef]
- Zonia, L.E.; Stebbins, N.E.; Polacco, J.C. Essential role of urease in germination of nitrogen-limited Arabidopsis thaliana seeds. Plant Physiol. 1995, 107, 1097–1103. [Google Scholar] [CrossRef]
- Zhengping, W.; Van Cleemput, O.; Demeyer, P.; Baert, L. Effect of urease inhibitors on urea hydrolysis and ammonia volatilization. Biol. Fertil. Soils 1991, 11, 43–47. [Google Scholar] [CrossRef]
- Li, W.Y.; Ni, W.W.; Ye, Y.X.; Fang, H.L.; Pan, X.M.; He, J.L.; Zhou, T.L.; Yi, J.; Liu, S.S.; Zhou, M.; et al. N-monoarylacetothioureas as potent urease inhibitors: Synthesis, SAR, and biological evaluation. J. Enzym. Inhib. Med. Chem. 2020, 35, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Aispuro-Pérez, A.; López-Ávalos, J.; García-Páez, F.; Montes-Avila, J.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Montaño, S.; Calderón-Zamora, L.; Osuna-Martínez, U.; et al. Synthesis and molecular docking studies of imines as α-glucosidase and α-amylase inhibitors. Bioorganic Chem. 2020, 94, 103491. [Google Scholar] [CrossRef] [PubMed]
- Hamad, A.; Khan, M.A.; Rahman, K.M.; Ahmad, I.; Ul-Haq, Z.; Khan, S.; Shafiq, Z. Development of sulfonamide-based Schiff bases targeting urease inhibition: Synthesis, characterization, inhibitory activity assessment, molecular docking and ADME studies. Bioorganic Chem. 2020, 102, 104057. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kitahara, F.; Nakamura, T.; Kojima, Y.; Fujino, M.A. Peptic ulcer in patients with diabetes mellitus. Nihon Rinsho. Jpn. J. Clin. Med. 2002, 60, 1580–1584. [Google Scholar]
- Tachecí, I.; Bures, J. Peptic ulcer disease in patients with diabetes mellitus. Vnitr. Lek. 2011, 57, 347–350. [Google Scholar]
- Rostom, S.A.; Badr, M.H.; Abd El Razik, H.A.; Ashour, H.M.; Abdel Wahab, A.E. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Arch. Der Pharm. 2011, 344, 572–587. [Google Scholar] [CrossRef]
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindy, S.M. Unveiling a versatile heterocycle: Pyrazoline–a review. RSC Adv. 2017, 7, 46999–47016. [Google Scholar] [CrossRef]
- Chandra, T.; Garg, N.; Lata, S.; Saxena, K.K.; Kumar, A. Synthesis of substituted acridinyl pyrazoline derivatives and their evaluation for anti-inflammatory activity. Eur. J. Med. Chem. 2010, 45, 1772–1776. [Google Scholar] [CrossRef]
- Karabacak, M.; Altıntop, M.D.; Çiftçi, H.İ.; Koga, R.; Otsuka, M.; Fujita, M.; Özdemir, A. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules 2015, 20, 19066–19084. [Google Scholar] [CrossRef]
- Kumar, K.A.; Govindaraju, M. Pyrazolines: Versatile molecules of synthetic and pharmaceutical applications-a review. Int. J. ChemTech Res 2015, 8, 313–322. [Google Scholar]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole ring—A biologically active scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef] [PubMed]
- Nehra, B.; Rulhania, S.; Jaswal, S.; Kumar, B.; Singh, G.; Monga, V. Recent advancements in the development of bioactive pyrazoline derivatives. Eur. J. Med. Chem. 2020, 205, 112666. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1, 3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Dawood, K.M.; Eldebss, T.M.; El-Zahabi, H.S.; Yousef, M.H. Synthesis and antiviral activity of some new bis-1,3-thiazole derivatives. Eur. J. Med. Chem. 2015, 102, 266–276. [Google Scholar] [CrossRef]
- Gollapalli, M.; Taha, M.; Javid, M.T.; Almandil, N.B.; Rahim, F.; Wadood, A.; Mosaddik, A.; Ibrahim, M.; Alqahtani, M.A.; Bamarouf, Y.A. Synthesis of benzothiazole derivatives as a potent α-glucosidase inhibitor. Bioorganic Chem. 2019, 85, 33–48. [Google Scholar] [CrossRef]
- Siddiqui, A.A.; Partap, S.; Khisal, S.; Yar, M.S.; Mishra, R. Synthesis, anti-convulsant activity and molecular docking study of novel thiazole pyridazinone hybrid analogues. Bioorganic Chem. 2020, 99, 103584. [Google Scholar] [CrossRef]
- Jaishree, V.; Ramdas, N.; Sachin, J.; Ramesh, B. In vitro antioxidant properties of new thiazole derivatives. J. Saudi Chem. Soc. 2012, 16, 371–376. [Google Scholar] [CrossRef]
- Kasralikar, H.M.; Jadhavar, S.C.; Goswami, S.V.; Kaminwar, N.S.; Bhusare, S.R. Design, synthesis and molecular docking of pyrazolo [3, 4d] thiazole hybrids as potential anti-HIV-1 NNRT inhibitors. Bioorganic Chem. 2019, 86, 437–444. [Google Scholar] [CrossRef]
- Modrić, M.; Božičević, M.; Faraho, I.; Bosnar, M.; Škorić, I. Design, synthesis and biological evaluation of new 1, 3-thiazole derivatives as potential anti-inflammatory agents. J. Mol. Struct. 2021, 1239, 130526. [Google Scholar] [CrossRef]
- Kesari, C.; Rama, K.R.; Sedighi, K.; Stenvang, J.; Björkling, F.; Kankala, S.; Thota, N. Synthesis of thiazole linked chalcones and their pyrimidine analogues as anticancer agents. Synth. Commun. 2021, 51, 1406–1416. [Google Scholar] [CrossRef]
- Rana, M.; Arif, R.; Khan, F.I.; Maurya, V.; Singh, R.; Faizan, M.I.; Yasmeen, S.; Dar, S.H.; Alam, R.; Sahu, A.; et al. Pyrazoline analogs as potential anticancer agents and their apoptosis, molecular docking, MD simulation, DNA binding and antioxidant studies. Bioorganic Chem. 2021, 108, 104665. [Google Scholar] [CrossRef] [PubMed]
- Tok, F.; Abas, B.İ.; Çevik, Ö.; Koçyiğit-Kaymakçıoğlu, B. Design, synthesis and biological evaluation of some new 2-Pyrazoline derivatives as potential anticancer agents. Bioorganic Chem. 2020, 102, 104063. [Google Scholar] [CrossRef] [PubMed]
- Ardiansah, B. Pharmaceutical importance of pyrazoline derivatives: A mini review. J. Pharm. Sci. Res. 2017, 9, 1958–1960. [Google Scholar]
- Matiadis, D.; Sagnou, M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, R.; Sadiq, A.; Alsantali, R.I.; Mughal, E.U.; Alsharif, M.A.; Naeem, N.; Javid, A.; Al-Rooqi, M.M.; Chaudhry, G.E.S.; Ahmed, S.A. Synthesis and evaluation of 1, 3, 5-triaryl-2-pyrazoline derivatives as potent dual inhibitors of urease and α-glucosidase together with their cytotoxic, molecular modeling and drug-likeness studies. ACS-Omega 2022, 7, 3775–3795. [Google Scholar] [CrossRef] [PubMed]
- Zelelew, D.; Endale, M.; Melaku, Y.; Kedir, F.; Demissie, T.B.; Ombito, J.O.; Eswaramoorthy, R. Synthesis, Antibacterial, and Antioxidant Activities of Thiazolyl-Pyrazoline Schiff Base Hybrids: A Combined Experimental and Computational Study. J. Chem. 2022, 2022, 3717826. [Google Scholar] [CrossRef]
- Hawash, M.M.; Kahraman, D.C.; Eren, F.; Atalay, R.C.; Baytas, S.N. Synthesis and biological evaluation of novel pyrazolic chalcone derivatives as novel hepatocellular carcinoma therapeutics. Eur. J. Med. Chem. 2017, 129, 12–26. [Google Scholar] [CrossRef]
- Khan, Y.; Khan, S.; Rehman, W.; Hussain, R.; Maalik, A.; Ali, F.; Khan, M.U.; Sattar, A.; Assiri, M.A. Hybrid Molecules of Thiadiazole-Based Benzothioate and Benzenesulfonothioate: Synthesis, Structural Analysis, and Evaluation as Potential Inhibitors of Thymidine Phosphorylase and β-Glucuronidase through In Vitro and In Silico Approaches. J. Mol. Struct. 2023, 136439. [Google Scholar] [CrossRef]
- Hussain, R.; Rehman, W.; Rahim, F.; Khan, S.; Alanazi, A.S.; Alanazi, M.M.; Rasheed, L.; Khan, Y.; Shah, S.A.A.; Taha, M. Synthesis, in vitro thymidine phosphorylase inhibitory activity and molecular docking study of novel pyridine-derived bis-oxadiazole bearing bis-schiff base derivatives. Arab. J. Chem. 2023, 16, 104773. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Rahim, F.; Hussain, R.; Khan, Y.; Khan, M.S.; Iqbal, R.; Ali, B.; Albeshr, M.F. Synthesis, biological evaluation and molecular docking study of pyrimidine based thiazolidinone derivatives as potential anti-urease and anti-cancer agents. J. Saudi Chem. Soc. 2023, 27, 101688. [Google Scholar] [CrossRef]
- Zaman, K.; Rahim, F.; Taha, M.; Ullah, H.; Wadood, A.; Nawaz, M.; Khan, F.; Wahab, Z.; Shah, S.A.A.; Rehman, A.U.; et al. Synthesis, in vitro urease inhibitory potential and molecular docking study of Benzimidazole analogues. Bioorganic Chem. 2019, 89, 103024. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Iqbal, S.; Shah, M.; Rehman, W.; Khan, S.; Rasheed, L.; Rahim, F.; Dera, A.A.; Kehili, S.; Elkaeed, E.B.; et al. Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study. Molecules 2022, 27, 6457. [Google Scholar] [CrossRef]
- Hussain, R.; Rehman, W.; Rahim, F.; Khan, S.; Taha, M.; Khan, Y.; Sardar, A.; Khan, I.; Shah, S.A.A. Discovery of imidazopyridine derived oxadiazole-based thiourea derivatives as potential anti-diabetic agents: Synthesis, in vitro antioxidant screening and in silico molecular modeling approaches. J. Mol. Struct. 2023, 1293, 136185. [Google Scholar] [CrossRef]
- Saeedian Moghadam, E.; Al-Sadi, A.M.; Talebi, M.; Amanlou, M.; Amini, M.; and Abdel-Jalil, R. Novel benzimidazole derivatives; synthesis, bioactivity and molecular docking study as potent urease inhibitors. DARU J. Pharm. Sci. 2022, 30, 29–37. [Google Scholar] [CrossRef] [PubMed]
