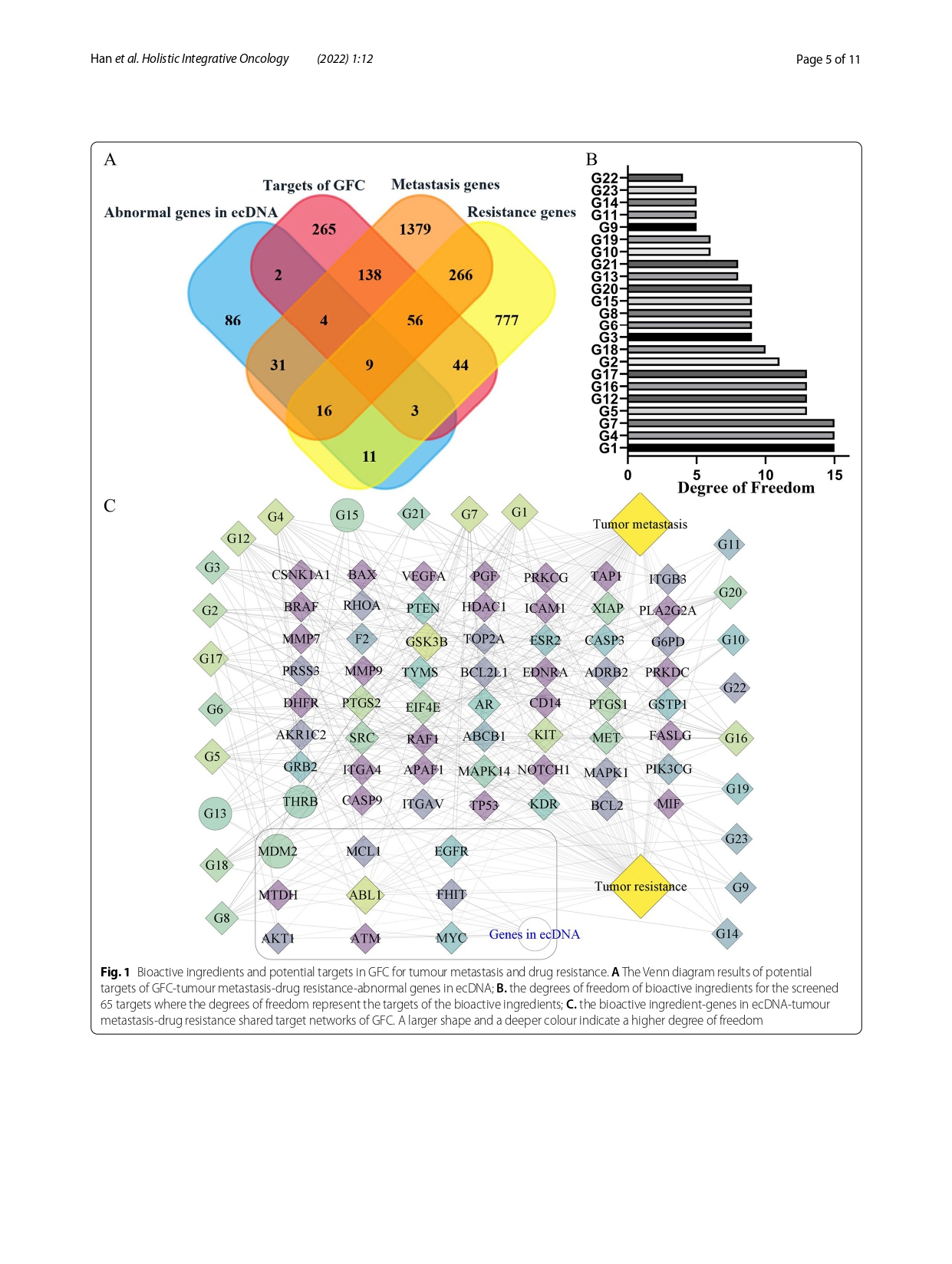
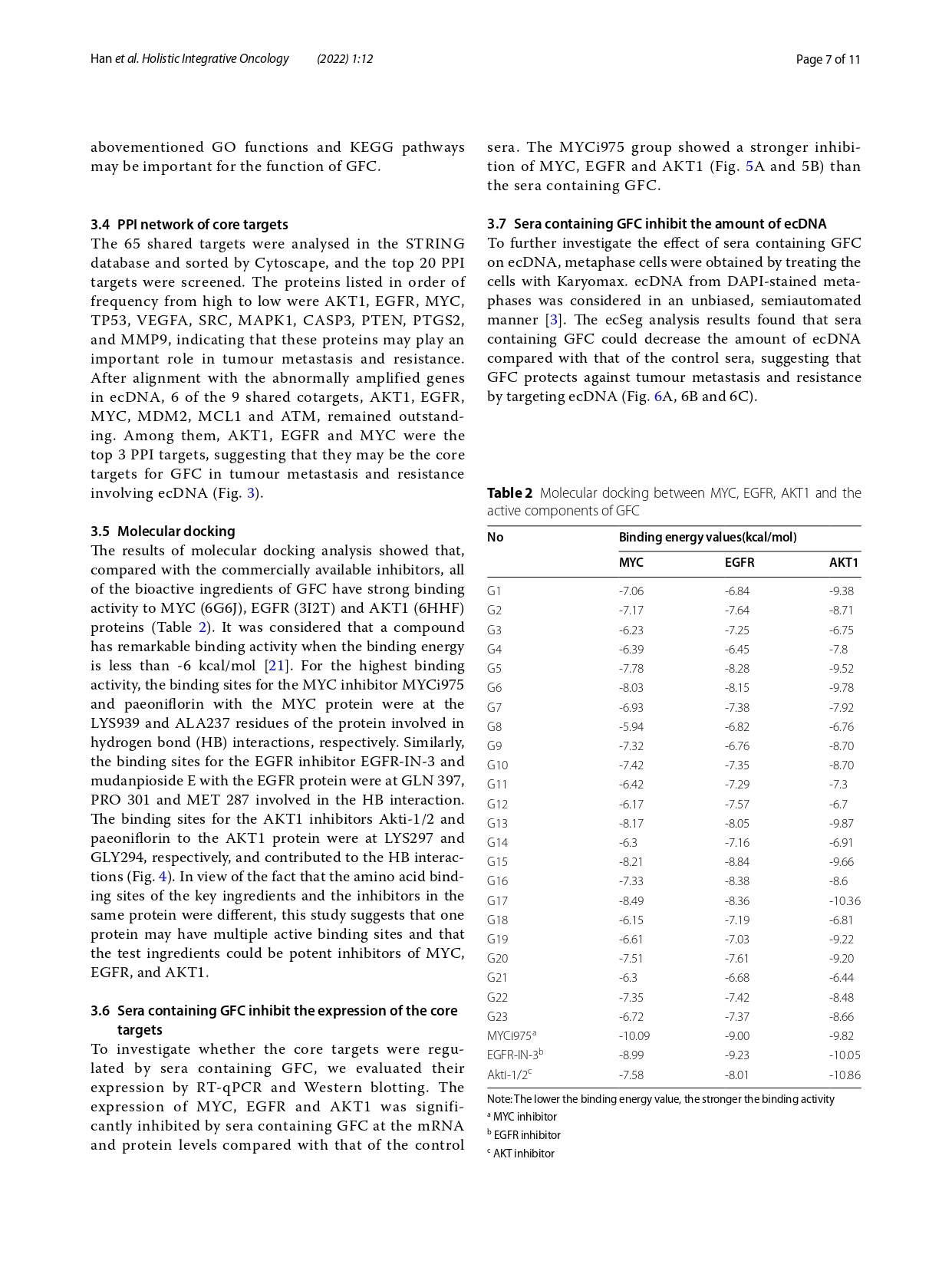
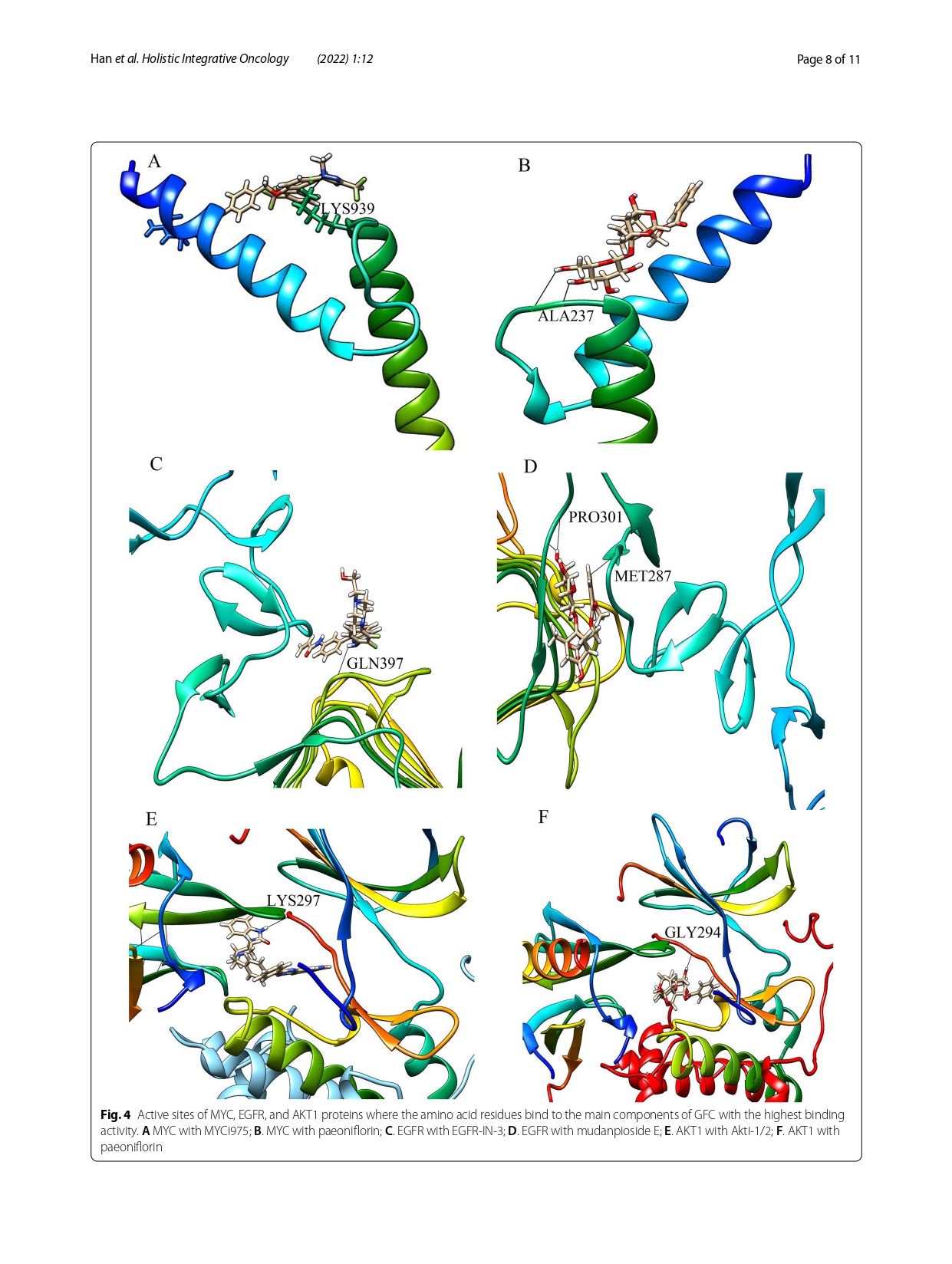
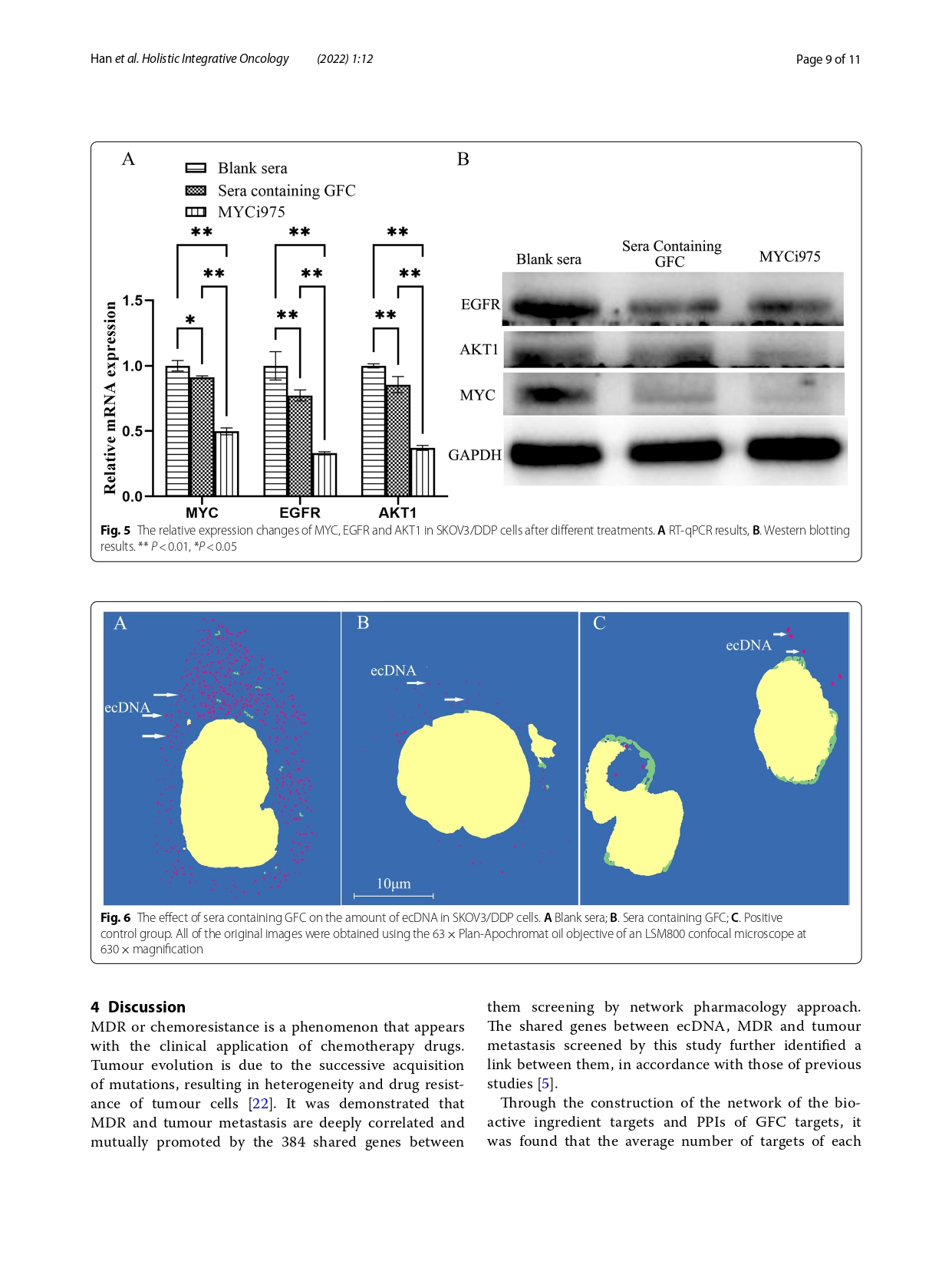
Knowledge
Dengue virus (DENV) is a major mosquito-borne human pathogen in tropical countries; however, there are currently no targeted antiviral treatments for DENV infection. Compounds 27 and 29 have been reported to be allosteric inhibitors of DENV RdRp with potent inhibitory effects. In this study, the structures of compounds 27 and 29 were optimized using computer-aided drug design (CADD) approaches. Nine novel compounds were synthesized based on rational considerations, including molecular docking scores, free energy of binding to receptor proteins, predicted Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) parameters, structural diversity, and feasibility of synthesis. Subsequently, the anti-DENV activity was assessed. In the cytopathic effect (CPE) assay conducted on BHK-21 cells using the DENV2 NGC strain, both SW-b and SW-d demonstrated comparable or superior activity against DENV2, with IC50 values of 3.58 ± 0.29 μM and 23.94 ± 1.00 μM, respectively, compared to that of compound 27 (IC50 = 19.67 ± 1.12 μM). Importantly, both SW-b and SW-d exhibited low cytotoxicity, with CC50 values of 24.65 μmol and 133.70 μmol, respectively, resulting in selectivity indices of 6.89 and 5.58, respectively. Furthermore, when compared to the positive control compound 3′-dATP (IC50 = 30.09 ± 8.26 μM), SW-b and SW-d displayed superior inhibitory activity in an enzyme inhibitory assay, with IC50 values of 11.54 ± 1.30 μM and 13.54 ± 0.32 μM, respectively. Molecular dynamics (MD) simulations elucidated the mode of action of SW-b and SW-d, highlighting their ability to enhance π–π packing interactions between benzene rings and residue W795 in the S1 fragment, compared to compounds 27 and 29. Although the transacylsulphonamide fragment reduced the interaction between T794 and NH, it augmented the interaction between R729 and T794. In summary, our study underscores the potential of SW-b and SW-d as allosteric inhibitors targeting the DENV NS5 RdRp domain. However, further in vivo studies are warranted to assess their