


1. Introduction
The development of molecules able to interfere with microbial infections was one of the most important achievements of modern medicine, but, nowadays, the dissemination of Antimicrobial Resistance (AMR) is heavily threatening this important success [1]. During 2019 alone, 1.2 million people died as a direct consequence of AMR, making AMR a leading cause of death worldwide, with a mortality rate higher than malaria and HIV [2].
The ESKAPE pathogens include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species, whose initials make the ESKAPE acronym. These are considered particularly worrying due to their increasing resistance towards several antimicrobials [2]. Among them, S. aureus deserves special mention, as it is responsible for severe nosocomial infections, significantly increasing the time of hospitalization, costs, and mortality. Recently, S. aureus has also become resistant to vancomycin and several other last-resort drugs, worsening the overall situation [3].
As suggested by the World Health Organization, one of the best potential strategies to overcome AMR is the development of molecules able to bind to new molecular targets that have not yet exploited by marketed antibiotics. In this regard, the bacterial cell division cycle is well known as a very promising target: as most bacteria divide through binary fission, the cell division machinery is highly conserved among different species, providing the potential for broad-spectrum agents [4,5]. FtsZ, a tubulin-like protein, is a key factor for organizing the cell division process. It initially polymerizes into a “Z ring” at the center of the cell. This first step is followed by the recruitment of other important cell division proteins that assemble the mature divisome, which, in turn, synthesizes the division septum that splits the original cell into two daughter cells [6,7]. FtsZ and the Z ring are crucial for this process, as inhibition of FtsZ polymerization or dynamics results in failed division, cell filamentation, and lysis. Several structurally distinct small-molecule inhibitors of FtsZ have been studied and developed over the last 15 years. Among them, the benzamide class is the most studied, thanks to its chemical accessibility, relatively low cytotoxicity, and positive results obtained with prototypes, such as 2,6-difluoro-3-nonyloxybenzamide and PC190723 (Figure 1) [8]. In particular, PC190723 strongly inhibits S. aureus FtsZ function in cell division, with a Minimal Inhibitory Concentration (MIC) of 1 μg/mL towards both methicillin-sensitive Staphylococcus aureus (MSSA) and methicillin-resistant S. aureus (MRSA) [8].
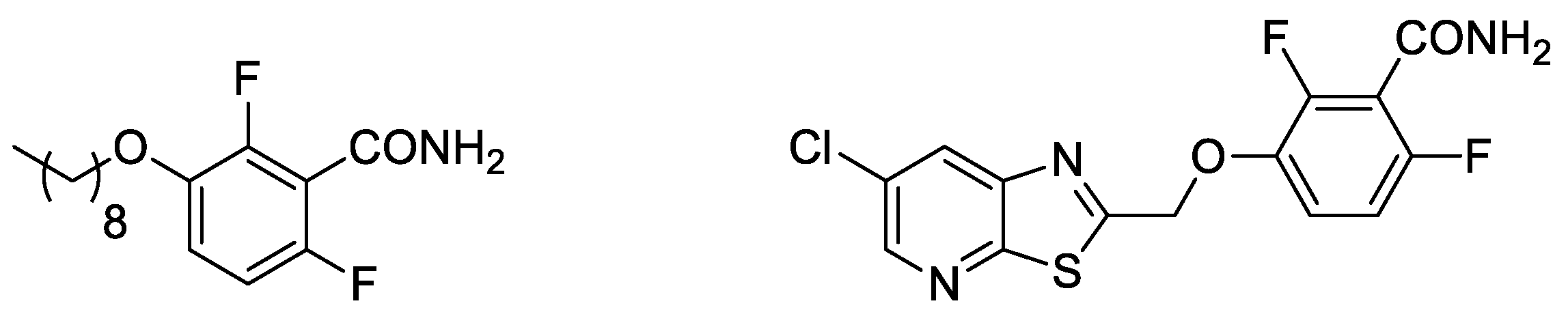
Figure 1. Structures of 2,6-difluoro-3-nonyloxybenzamide (left) and PC190723 (right).
Starting from these molecules, we recently first replaced the thiazolopyridine with an unsubstituted 1,4-benzodioxane ring, giving rise to FZ14 (Figure 2) [9], and then to several differently functionalized 1,4-benzodioxanes [10,11,12,13,14], resulting in the benzodioxane–benzamide FtsZ inhibitors class. The versatility of the 1,4-benzodioxane ring, a well-known scaffold in medicinal chemistry, led to the simple introduction of several structural modifications. Development of a robust and reliable computational model [13] provided the impetus to lengthen the methylenoxy to an ethylenoxy linker (FZ14 to FZ88), which resulted in strongly increasing the antimicrobial activity (Figure 2) [12]. A comprehensive Structure Activity Relationship (SAR) study led to the development of very potent compounds [14], named FZ95 and FZ100 (Figure 2), in which the benzamide pharmacophoric structure is linked to a naphthodioxane or a 5,6,7,8-tetrahydronaphthodioxane ring by an ethylenoxy linker.
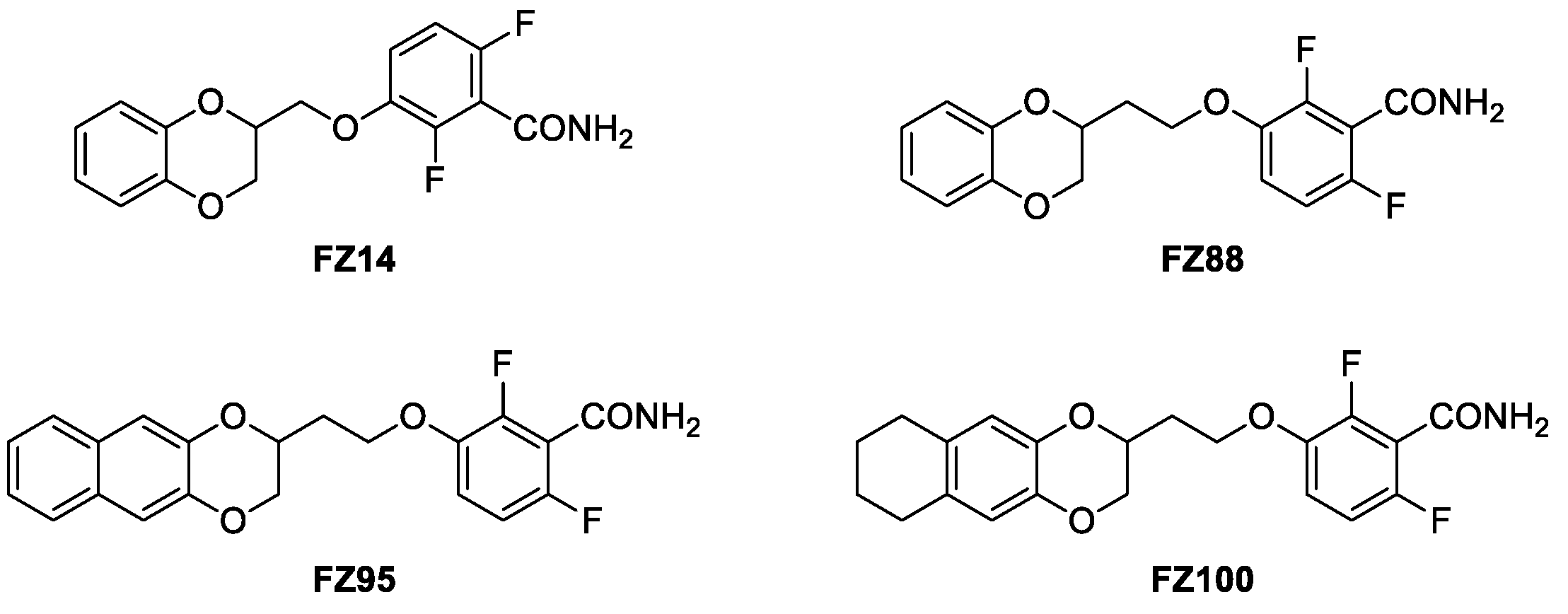
Figure 2. Compound FZ14, progenitor of the benzodioxane benzamide FtsZ inhibitors class; compound FZ88, its improved homologue, and compounds FZ95 and FZ100, presently the strongest derivatives of the series.
FZ95 and FZ100 are characterized by having very high potencies against both MRSA and MSSA, with MICs of 0.25 μg/mL (0.6 μM) and 0.1 μg/mL (0.25 μM), respectively. They also showed MICs under 0.1 μg/mL vs. B. subtilis, and their activity is accompanied by low to no cytotoxicity towards human MRC-5 cells [14]. In addition, we recently demonstrated how both of these compounds are able to interact with E. coli FtsZ and to interfere with its essential properties, therefore exerting antimicrobial activity on efflux-pump-defective E. coli strains, such as N43 (ΔacrAB) and ΔtolC [15]. These optimal microbiological profiles make these compounds very intriguing as potential antibiotics. Nevertheless, FZ95 and FZ100 have high lipophilicity, which will very likely translate to poor water solubility, poor oral bioavailability, a tendency to bioaccumulate, and other undesired pharmacokinetic properties. For both PC190723 and TXA707, which are characterized by similar problems, prodrugs have been designed by exploiting the benzamide group and converting it into a labile imide moiety [16]. Moreover, Stokes and collaborators, while working on the development of bromo-oxazole benzamides as FtsZ inhibitors [17,18], demonstrated how the introduction of hydroxy-methyl in the methylene bridge of the molecule resulted in an almost 10-fold increase in solubility without diminishing the antimicrobial activity. Moreover, the further derivatization with a liable succinic ester moiety redoubled the solubility value [17,18]. Starting from these findings, we continued the development of naphthodioxane–benzamides as FtsZ inhibitors by focusing on improving the physical–chemical properties. As a result, we recently designed an innovative set of derivatives characterized by an additional -OH on the linker between the two main moieties. This modification introduces a second stereogenic center, prompting us to isolate and individually evaluate the erythro and threo isomers. As reported in our recent work focused on E. coli as a target, FZ116 (Figure 3) resulted in very strong in vitro inhibition of E. coli FtsZ and inhibition of cell division in efflux-pump-defective E. coli [15]. Stokes and co-workers also observed how the introduction of a methyl group in the same position resulted in the enhancement of the antimicrobial activity vs. S. aureus without positively affecting the solubility [17]. These results suggest how both hydrophilic and lipophilic substituents could be tolerated.
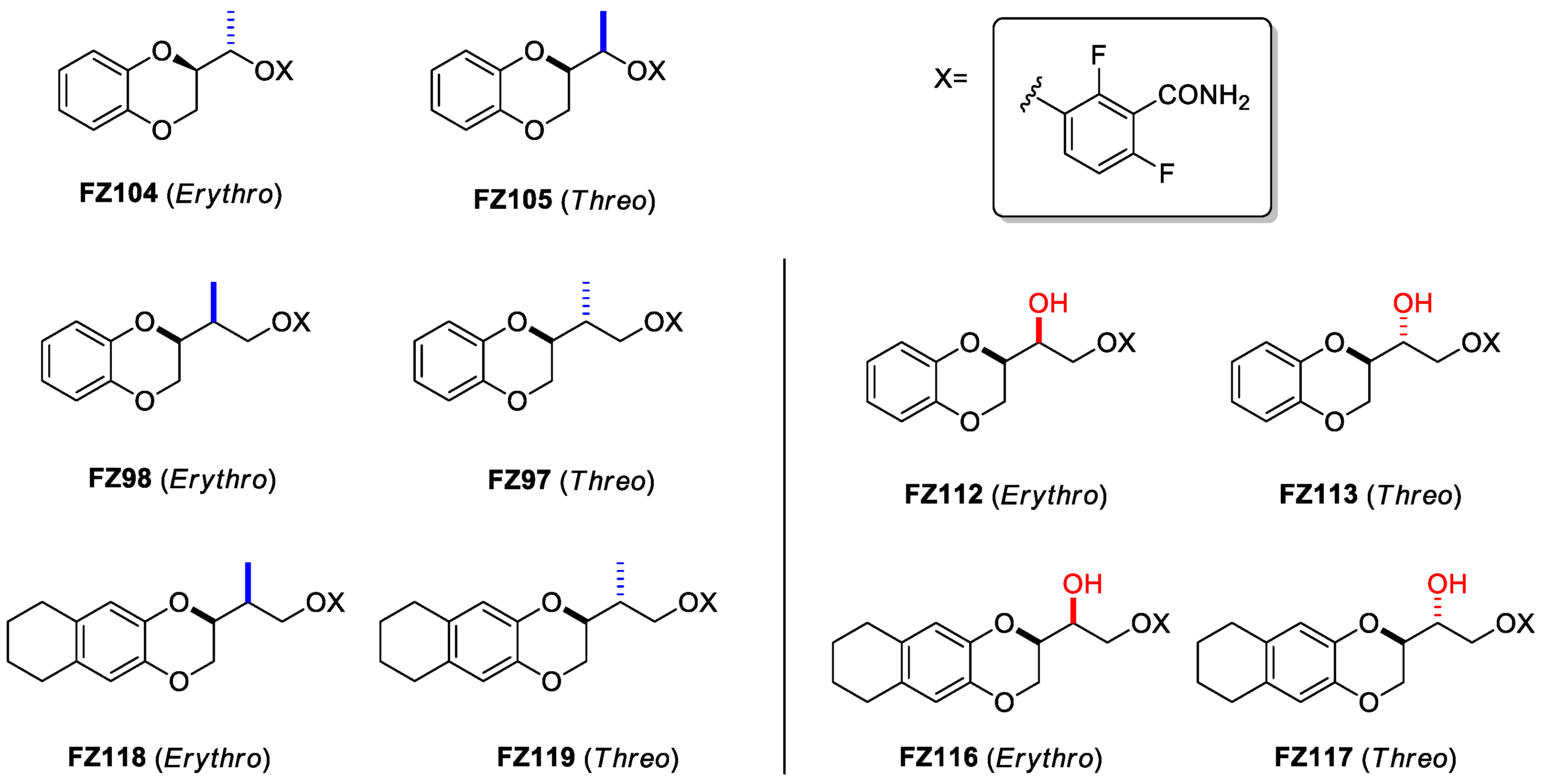
Figure 3. Structures of compounds FZ104, FZ105, FZ98, FZ97, FZ118 and FZ119, bearing the methyl substituent (highlighted in blue), and of compounds FZ112, FZ113, FZ116 and FZ117, bearing the hydroxy substituent (highlighted in red), which are the focus of the present study.
Starting from these considerations, in this work, we focused our attention on the effects of introducing different linker substituents on the Gram-positive antimicrobial activity of these compounds. We firstly determined how introducing a methyl group on the structures of FZ14, FZ88, and FZ100 influenced the antimicrobial activity. As the introduction of a methyl group resulted in a second stereogenic center, we thus characterized each erythro and threo isomer (derivatives FZ104, FZ105, FZ98, FZ97, FZ118, and FZ119 in Figure 3). In addition, we evaluated the effects of each novel compound, as well as those of FZ116, FZ117, FZ112, and FZ113 (Figure 3), which were previously described as bearing the -OH group as a substituent, as antimicrobials against Gram-positive S. aureus and B. subtilis.
2. Results
2.1. Chemistry
Scheme 1 reports the synthetic pathway for achieving compounds FZ104 and FZ105, which started from methyl ketone 1, an intermediate developed by our research team and whose preparation was previously described and applied [19]. The methyl ketone 1 underwent reduction (2) and further mesylation, thus obtaining compound 3, which was substituted with the pharmacophoric 2,6-difluoro-3-hydroxybenzamide (in house prepared [9]). The separation of the erythro and the threo isomers was achieved using flash chromatography on silica gel, although further synthetic work was required for the identification of the correct isomer. We therefore synthesized the pure (S,R) erythro enantiomer, as described in Scheme 2.
We started with the commercially available D-Mannitol diacetonide, which was converted into the aldehyde 4 through the oxidative C-C bond cleavage using NaIO4. This intermediate was then reduced (5), and the hydroxylic function was benzylated (6). The two isomers were then isolated through flash chromatography, and the (R,S) stereoisomer was identified by comparing its 1H-NMR spectrum in CDCl3 and its optical properties with the limited data from the literature [20,21]. The synthesis then continued with the (R,S) isomer through the hydrolysis of the acetonide, accomplishing intermediate 7 (a comparison of 1H-NMR chemical shifts with a different literature reference [22] allowed the further confirmation of its absolute configuration) and mesylation of both the hydroxy functions (8). These were rapidly substituted with catechol, achieving the 1,4-benzodioxane ring with a complete inversion of the configuration of the benzodioxane stereocenter (9). The final debenzylation (10), mesylation (11), and substitution with the pharmacophoric 2,6-difluoro-3-hydroxybenzamide allowed for the obtainment of enantiopure (S,R)-FZ105.
To obtain compounds FZ98 and FZ97 (Scheme 3), the synthesis started from derivative 1, which underwent the Wittig reaction (12) and anti-Markovnikov water addition (13). Following the strategy described for FZ104 and FZ105, the hydroxy function was mesylated (14) and substituted with the pharmacophoric benzamide. At this point, the two isomers were isolated on preparative HPLC, and their configuration was easily assessed through comparison of 1H-NMR spectra and chromatographic data, therefore defining FZ98 as erythro and FZ97 as threo.
The syntheses of derivatives FZ118 and FZ119 followed a complete novel synthetic strategy, as reported in Scheme 4. After having applied an in house previously described synthetic strategy [23] and having achieved the 5,6,7,8-tetrahydronaphthalen-2,3-diol, this was reacted with methyl 3,4-dibromo-2-methylbutanoate (17), which was obtained through deconjugative transposition (15), bromination (16), and esterification of the commercially available tiglic acid. The nature of the derivative obtained allowed for the separation of the two isomers using flash chromatography and the definition of the erythro from the threo isomer through comparison of 1H-NMR spectra. The syntheses proceeded individually for the two isomers through reduction (19, 20), mesylation (21, 22), and condensation with the pharmacophoric 2,6-difluorobenzamide, therefore achieving compounds FZ118 and FZ119.
2.2. Antimicrobial Activity on Staphylococcus aureus
As described in our previous work [12,13,14], we evaluated all the compounds mentioned above on different S. aureus strains. We tested on both methicillin-sensitive S. aureus (MSSA, ATCC29213) and two clinical isolates of S. aureus showing multi-drug resistance (MDRSA). MDRSA 12.1 is resistant towards cefoxitin, gentamicin, kanamycin, rifampicin, streptomycin, sulfamethoxazole, and tetracycline, while MDRSA 11.7 shows resistance to cefoxitin, ciprofloxacin, clindamycin, erythromycin, quinupristin-dalfopristin association, tetracycline, tiamulin, and trimethoprim. We analyzed the MICs and, if the antibacterial activity was promising, we also determined the percentage of cytotoxicity using the MTT assay on human MRC-5 cells and reported it as TD90. The results of antibacterial properties of all of the synthesized derivatives are shown in , and they are compared with their reference compounds FZ14, FZ88, and FZ100.
As observed, neither the introduction of the methyl nor of the hydroxy led to a further strong increase in the antimicrobial activity vs. S. aureus. Nonetheless, FZ116, the erythro hydroxy derivative of FZ100, and FZ118 and FZ119, the methyl derivatives of FZ100, retain promising activity versus all of the bacterial strains and a negligible degree of cytotoxicity.
2.3. Antimicrobial Activity on Bacillus subtilis and Effects of FZ116 and FZ117 on Its Inhibition
We further evaluated the most promising compounds on B. subtilis, a model Gram-positive species used previously in evaluating other benzamides [24], including by our research group [14]. The B. subtilis strain WM5126 expresses a xylose-inducible GFP-tagged ZapA protein. As ZapA interacts directly with FtsZ, GFP-ZapA serves as a proxy for the Z ring without significantly altering the properties of the FtsZ target itself. We first tested the effects of the compounds on colony viability on solid medium (Figure 4). Although none of the compounds were as potent as FZ100, FZ116 and FZ119 killed B. subtilis at the lowest doses, with FZ117 and FZ118 being less effective (Figure 4A). Among the less potent compounds, FZ104 was the best, as it was able to kill at 6 µg/mL (~20 µM), whereas FZ98, FZ112, and FZ113 still permitted growth at this concentration (Figure 4B). MIC values from microdilution experiments, shown in the right column in Figure 4, largely mirrored the spot viability data. The effects of the compounds on B. subtilis also generally tracked with their effects on S. aureus, with FZ100 being the most potent, followed by FZ116, FZ119, and FZ118, and with FZ112, FZ113, and FZ98 having the lowest antimicrobial activities.
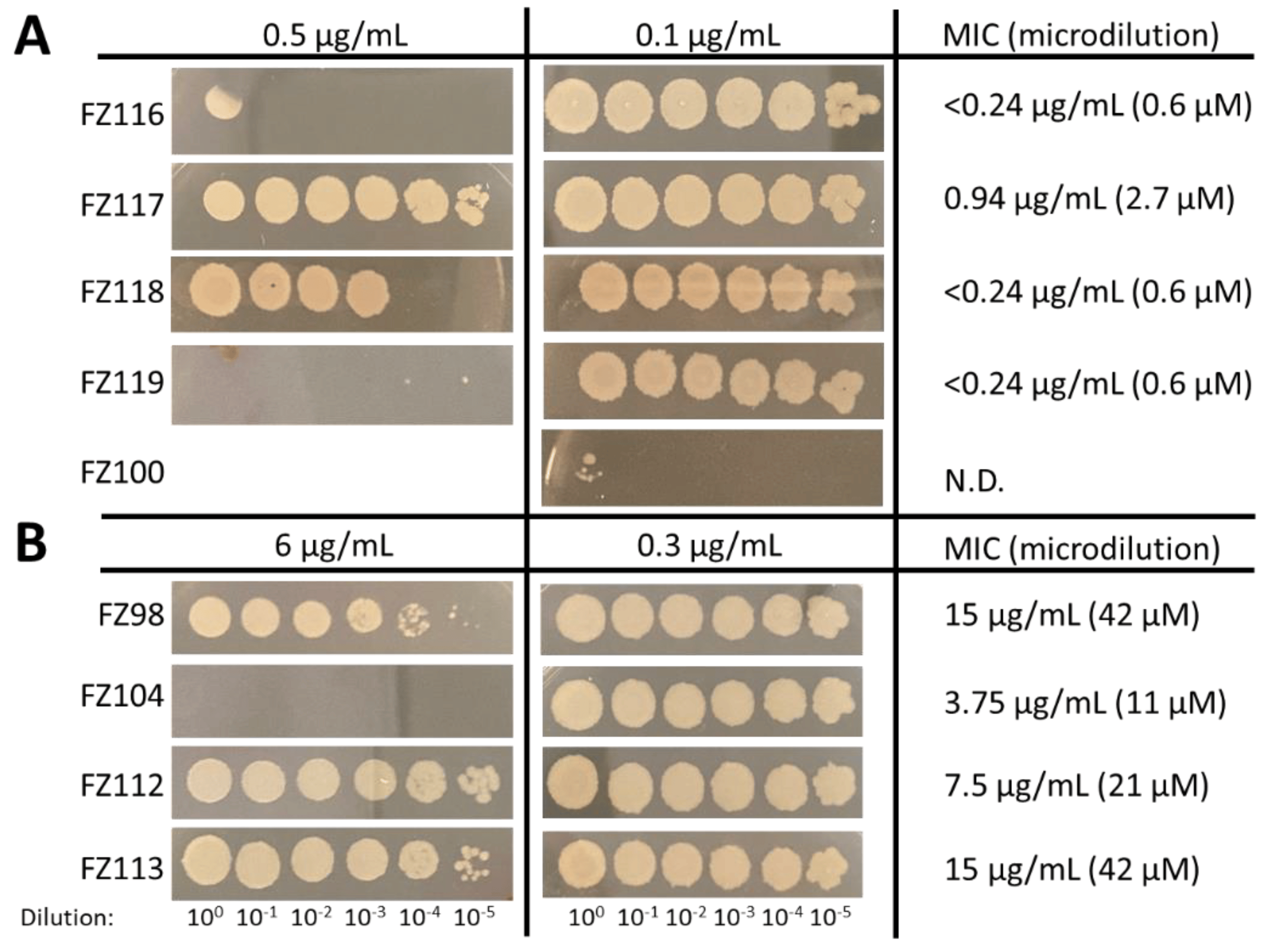
Figure 4. Effects of the compounds on viability of B. subtilis strain WM5126, showing spot dilutions of cultures on agar medium (left) and MICs from broth microdilutions (right) in the presence of the more potent (A) and less potent (B) compounds, as compared with the previously characterized FZ100. N.D.: not determined.
To test if the differential effects of FZ116 and FZ117 enantiomers could be detected as FtsZ assembly changes in B. subtilis cells, we visualized GFP-ZapA patterns after growth in inhibitory concentrations of FZ116 and FZ117 (0.3 µg/mL or 0.8 µM). Cells grown with no added compound grew as typical single bacilli and short chains, with well- defined Z rings clearly visible (Figure 5, left panel, single yellow arrows). In contrast, FtsZ rings were largely disrupted in cells with FZ116, with most of the fluorescence localized to speckles (middle panel, forked arrows) with only rare intact Z rings (yellow arrows). The lower potency of FZ117 was manifested by a mixture of FtsZ speckles and Z rings, although, as with FZ116-treated cells, most of the cells were filamentous because of division defects. This conversion of FtsZ rings into a speckle pattern was observed previously, both with FZ100 [14] and with the original benzamide derivatives [24].
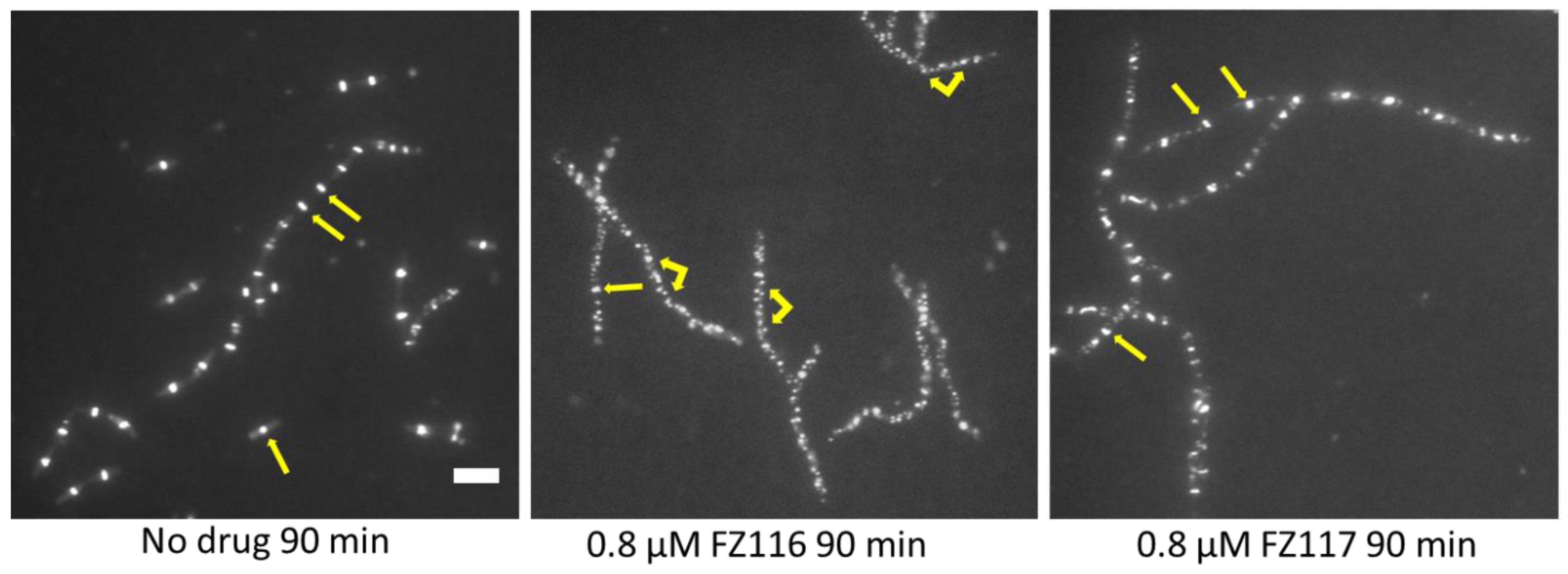
Figure 5. Effects of the compounds on FtsZ assembly and patterning in live B. subtilis WM 5126 cells expressing GFP-ZapA as a proxy for FtsZ. Arrows highlight normal FtsZ rings, both in single dividing cells (left panel), chains of dividing cells (left panel), and nondividing filamentous cells (middle and right panels). Forked arrows highlight FtsZ speckles that result from the disruption of FtsZ rings by FZ116 and, to a lesser extent, by the same concentration of the comparatively less potent compound FZ117. Scale bar, 5 μm.
2.4. Computational Studies
2.5. Linker Effect about the Accomodation into the Binding Pocket
To understand the importance and the role of OH and CH3 substituents, especially on the influence of the molecule accommodation into the binding site, we analyzed all of the possible aminoacidic residues in the radius of 3 Å around the poses able to form the three key HBs. This allowed us to identify two different clusters, depending on the aminoacidic environment and, consequently, on the potential additional interactions . The former group is characterized by the clear and definite binding interaction with the residue Thr309 (Figure 7A) via a HB, and the latter by the absence of residues around the substituent (Figure 7B). In addition, two peculiar patterns should be defined between the erythro and threo isomers bearing the OH substituent, although the presence of a weak HB interaction with Thr309 was prevalent in the threo-OH group.
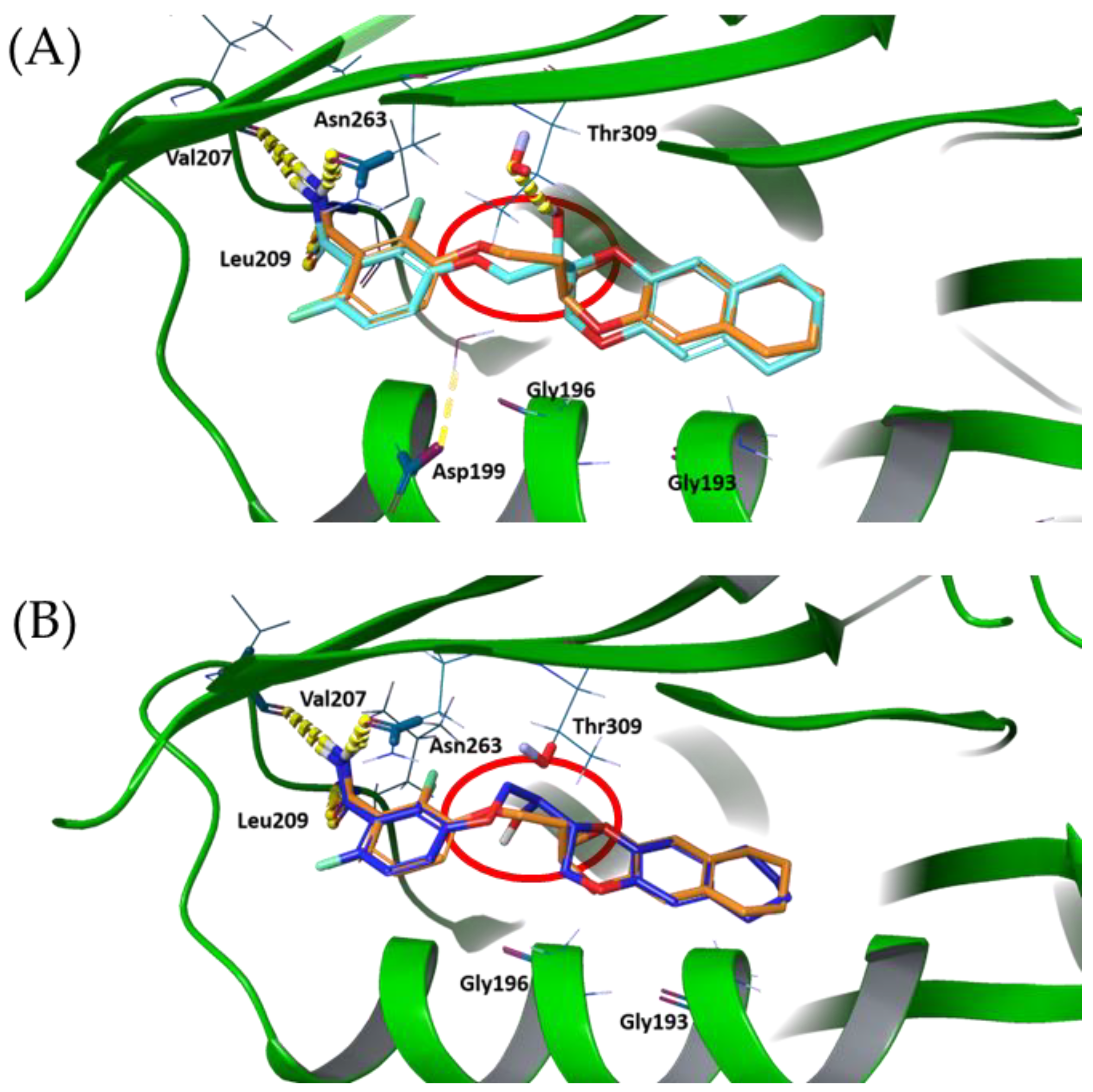
Figure 7. FZ117 pose (A) and FZ116 pose (B), superimposed with FZ100 (orange coloured). Hydrogen bonds are highlighted in yellow to be clearly visible.
Finally, we superposed FZ100 with the two new inhibitors clusters, therefore allowing us to identify a novel and peculiar behavior involving the binding mode of the benzodioxane moiety. It is interesting to highlight that all poses, except for FZ104, show a 180º rotation of the benzodioxane moiety into the hydrophobic binding subpocket driven by the ethylenoxy linker ( and ). This highlights that the binding and conformation are driven by the three key HB interactions in the benzamide pocket and the lipophilic tail in the hydrophobic subpocket. Therefore, the linker acts as a spacer to accommodate the key pharmacophoric features in the molecule.
This unexpected harboring is easily visible in Figure 7, where the red circles highlight the twisting of the linker, thus allowing the 180° rotation of the tetrahydronaphthodioxane ring plane around its axis of symmetry.
3. Discussion
The evaluation of antimicrobial assays on both S. aureus and B. subtilis, together with the predicted properties, the docking poses, and the scores, led us to several interesting conclusions.
First, the development of the differentially substituted FZ116, FZ117, FZ118 and FZ119, the tetrahydro naphthodioxane compounds, allowed us to obtain promising derivatives with antimicrobial activities against both S. aureus and B. subtilis. This consideration, together with the recent results with efflux-deficient E. coli strains [15], strongly pave the way to FtsZ inhibitors able to possess a wide spectrum of action not restricted to exclusively Gram-positive or Gram-negative strains.
Different hypotheses should be tested depending on the substituent inserted on the ethylenoxy linker, because methyl or hydroxy substituents in turn led to different outcomes, both in terms of antimicrobial potencies and computational properties.
Alternately, the introduction of a methyl group does not result in any possible additional interaction with aminoacidic residues of the protein to further strengthen the harboring into the binding site; consequently, this did not lead to an increase in the antimicrobial activity. The main effect of its insertion is in terms of spatial volume, because this functional group is more hindered than the hydroxy function and spatially spherical, without any possibility to be oriented by the aminoacidic environment around it. As a result, no differences between erythro and threo isomers in terms of antimicrobial activities could be detected.
A completely different outcome was found for the OH derivatives, because several possible aminoacidic residues, especially Threonine 309, are present within the binding site and could allow for the establishment of additional hydrogen bonds. Moreover, the hydroxy group is less hindered and, more importantly, it is angular and it could be differently oriented, as it could be driven by surrounding residues.
The results achieved in this work are perfectly in line with the computational model developed so far [12,13,14]. The model suggested to us how the FtsZ binding subpocket, responsible for harboring the benzodioxane or tetrahydronaphthodioxane, is characterized by hydrophobic and non-aromatic residues and how the linker between the two aromatic scaffolds is deputed to the best accommodation into this hydrophobic cavity.
Here, we further demonstrated how the linker is not involved in specific interactions and how it is able to freely orientate itself to achieve the best accommodation of the naphthodioxane moiety. Moreover, considering the symmetric and linear nature of the tetrahydronaphthodioxane, the degree of freedom is even higher, with the possibility of performing a 180° rotation around the axis of symmetry to maximize the interaction within the protein binding pocket. Finally, focusing on the best computational poses of FZ116 and FZ117, in both of the isomers, it seems that the 180° rotation induces the OH group orientation in order to have the polar substituent out of the binding pocket and available for further derivatization, such as the preparation of prodrugs or probes.
4. Materials and Methods
4.1. Chemistry
The totality of the reagents and the solvents were purchased from commercial sources (Merck KGaA (Darmstadt, Germany), Fluorochem (Hadfield, United Kingdom), and Carlo Erba (Cornaredo, Milan, Italy)), and they were used without further purification or distillation.
Silica gel matrix was used in both TLC (thin-layer chromatography on aluminum foils having a fluorescent indicator 254 nm) and in flash chromatography (particle size 230–400 mesh, Carlo Erba) on Puriflash XS 420 (Sepachrom Srl, Rho (MI), Italy). The visualization was performed with UV light at 254 nm or at 280 nm (λ).
A Varian (Palo Alto, CA, USA) Mercury 300 NMR spectrometer/Oxford Narrow Bore superconducting magnet operating at 300 MHz was used for all 1H-NMR spectra. 13C-NMR spectra were acquired at 75 MHz. The chemical shifts are reported in ppm (δ), relative to the residual solvent as an internal standard. The following abbreviations refer to signal multiplicity: s = singlet, d = doublet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dq = doublet of quadruplets, quint = quintet, m = multiplet, bs = broad singlet.
The final products (FZ104, FZ105, FZ97, FZ98, FZ118, and FZ119) were analyzed through reverse-phase HPLC using a Waters XBridge C-18 column (5 µm, 4.6 mm × 150 mm) on an Elite LaChrom HPLC system (Hitachi, San Jose, CA; USA) equipped with a DAD (diode array detector). HPLC methods used two different solvents: A: H2O with 0.10% TFA, and B: acetonitrile with 0.10% TFA. Method A: isocratic elution 60% A and 40% B with 30 min run time and a flow rate of 1 mL/min. Method B: isocratic elution 70% A and 30% B with 40 min run time and a flow rate of 1.2 mL/min. Method C: gradient elution from 90% A to 10% A in 25 min with 35 min run time and a flow rate of 1 mL/min. The purities of the final products was quantified at specific λ values, depending on the compound, and all were >95%. The relative retention times are reported in each experimental section. The melting points were determined through DSC analysis using a DSC 1020 apparatus (TA Instruments, New Castle, DE, USA).
The 1H- and 13C-NMR spectra of all of the final compounds, together with their HPLC profiles, are included in the .
4.2. Cells
Normal human lung fibroblasts (MRC-5) were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/mL penicillin, and 100 mg/mL streptomycin.
Gram-positive methicillin-sensitive Staphylococcus aureus (MSSA, ATCC 29213) was grown in Luria–Bertani broth (LB), as already described in our papers [11,12,13,14].
4.3. Antibacterial Activity
4.4. Thiazolyl Blue Tetrazolium Bromide (MTT) Cytotoxicity Assay
Compounds showing promising antibacterial activity were serially diluted in DMEM and tested for cytotoxicity on MRC-5 cells using the MTT assay (Sigma, St Louis, MO, USA), following the same protocol we previously reported [11,12,13,14].
4.5. Computational Studies
References
- Urban-Chmiel, R.; Marek, A.; Stępień-Pyśniak, D.; Wieczorek, K.; Dec, M.; Nowaczek, A.; Osek, J. Antibiotic Resistance in Bacteria-A Review. Antibiotics 2022, 11, 1079. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Holmes, N.E.; Johnson, P.D.R.; Howden, B.P. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J. Clin. Microbiol. 2012, 50, 2548–2552. [Google Scholar] [CrossRef]
- Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ. Antibiotics 2020, 9, 69. [Google Scholar] [CrossRef]
- Pradhan, P.; Margolin, W.; Beuria, T.K. Targeting the Achilles Heel of FtsZ: The Interdomain Cleft. Front. Microbiol. 2021, 12, 732796. [Google Scholar] [CrossRef]
- Cameron, T.A.; Margolin, W. Insights into the assembly and regulation of the bacterial divisome. Nat. Rev. Microbiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Haeusser, D.P.; Margolin, W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol. 2016, 14, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; de Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide Inhibitors of Bacterial Cell Division Protein FtsZ: Design, Synthesis, and Structure-Activity Relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef]
- Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; de Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics 2020, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. Benzodioxane-Benzamides as Antibacterial Agents: Computational and SAR Studies to Evaluate the Influence of the 7-Substitution in FtsZ Interaction. ChemMedChem 2020, 15, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Sebastián-Pérez, V.; Suigo, L.; Margolin, W.; Casiraghi, A.; Hrast, M.; Zanotto, C.; Zdovc, I.; Radaelli, A.; Valoti, E. Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics 2021, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Suigo, L.; Monterroso, B.; Sobrinos-Sanguino, M.; Alfonso, C.; Straniero, V.; Rivas, G.; Zorrilla, S.; Valoti, E.; Margolin, W. Benzodioxane-benzamides as promising inhibitors of Escherichia coli FtsZ. Int. J. Biol. Macromol. 2023, 253, 126398. [Google Scholar] [CrossRef]
- Rosado-Lugo, J.D.; Sun, Y.; Banerjee, A.; Cao, Y.; Datta, P.; Zhang, Y.; Yuan, Y.; Parhi, A.K. Evaluation of 2,6-difluoro-3-(oxazol-2-ylmethoxy)benzamide chemotypes as Gram-negative FtsZ inhibitors. J. Antibiot. 2022, 75, 385–395. [Google Scholar] [CrossRef]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Chauhan, P.K.; Collins, I.; Davies, D.T.; Gavade, M.; Kumar, D.; Lancett, P.; Macdonald, R.; et al. Design, synthesis and structure-activity relationships of substituted oxazole-benzamide antibacterial inhibitors of FtsZ. Bioorg. Med. Chem. Lett. 2014, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Berry, J.; Collins, I.; Czaplewski, L.G.; Logan, A.; Macdonald, R.; Macleod, L.; Peasley, H.; et al. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob. Agents Chemother. 2013, 57, 317–325. [Google Scholar] [CrossRef]
- Straniero, V.; Casiraghi, A.; Fumagalli, L.; Valoti, E. How do reaction conditions affect the enantiopure synthesis of 2-substituted-1,4-benzodioxane derivatives? Chirality 2018, 30, 943–950. [Google Scholar] [CrossRef]
- Abushanab, E.; Vemishetti, P.; Leiby, R.W.; Singh, H.K.; Mikkilineni, A.B.; Wu, D.C.J.; Saibaba, R.; Panzica, R.P. The chemistry of L-ascorbic and D-isoascorbic acids. 1. The preparation of chiral butanetriols and -tetrols. J. Org. Chem. 1988, 53, 2598–2602. [Google Scholar] [CrossRef]
- Holý, A. Preparation and synthetic utilization of 3-(adenin-9-yl)-2-hydroxyalkanoic acids and their derivatives. Collect. Czech. Chem. Commun. 1984, 49, 2148–2166. [Google Scholar] [CrossRef]
- Mohapatra, D.K.; Reddy, D.S.; Reddy, G.S.; Yadav, J.S. Synthesis of the C-8-C-24 Fragment of Maltepolide C by Using a Tandem Di-hydroxylation/S N 2 Cyclization Sequence. Eur. J. Org. Chem. 2015, 2015, 5266–5274. [Google Scholar] [CrossRef]
- Bolchi, C.; Catalano, P.; Fumagalli, L.; Gobbi, M.; Pallavicini, M.; Pedretti, A.; Villa, L.; Vistoli, G.; Valoti, E. Structure-affinity studies for a novel series of homochiral naphtho and tetrahydronaphtho analogues of alpha 1 antagonist WB-4101. Bioorg. Med. Chem. 2004, 12, 4937–4951. [Google Scholar] [CrossRef]
- Adams, D.W.; Wu, L.J.; Czaplewski, L.G.; Errington, J. Multiple effects of benzamide antibiotics on FtsZ function. Mol. Microbiol. 2011, 80, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Ralston, K.J.; Ramstadius, H.C.; Brewster, R.C.; Niblock, H.S.; Hulme, A.N. Self-Assembly of Disorazole C1 through a One-Pot Alkyne Metathesis Homodimerization Strategy. Angew. Chem. Int. Ed Engl. 2015, 54, 7086–7090. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: Glide; Schrödinger, LLC: New York, NY, USA, 2021.
- Schrödinger Release 2021-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021.
- Schrödinger Release 2021-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1: Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2021.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Maeda, Y.; Mizohata, E.; Inoue, T.; Kaul, M.; Parhi, A.K.; Lavoie, E.J.; Pilch, D.S.; Matsumura, H. Structural Flexibility of an Inhibitor Overcomes Drug Resistance Mutations in Staphylococcus aureus FtsZ. ACS Chem. Biol. 2017, 12, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Software Release 2021-1: Induced Fit Docking Protocol; Glide, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2021.
- QikProp 4.4 User Manual; Schrödinger Press LLC: New York, NY, USA, 2011.
- Schrödinger Release 2021-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2023.