


1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a disorder occurring in about 1 in every 400 to 1000 live births [1,2]. Most patients with ADPKD have an abnormality on chromosome 16 (PKD1 locus) [3], although other defects are reported [4,5]. In approximately eight percent of families, no mutation is detected. The disease leads to several cysts in the renal tubule, affecting its function over time. Renal ultrasonography is usually used for screening. A genetic diagnosis can be performed when a definitive diagnosis is required [6]. Usually, the renal manifestation is predominant: hematuria [7], proteinuria [8], nephrolithiasis [9], and finally renal insufficiency, although a prognostic model has been developed for identifying high-risk patients setting renal end-stage earlier [10]. Nevertheless, extra-renal manifestations such as cerebral aneurysms, hepatic and pancreatic cysts, and valvular abnormalities have also been reported [11,12,13,14]. Hypertension is another common disease present in these patients [15]. Still, although hypertension is common in most chronic progressive kidney diseases, the pathogenesis is somewhat different because it correlates to endothelial dysfunction, especially for a reduction in nitric oxide and an over-activation of the renin-angiotensin-aldosterone system [16,17,18]. Most patients with ADPKD die from cardiovascular causes [19,20,21]. An increase in arterial stiffness, left ventricular hypertrophy, and increased carotid intima-media thickness have been reported in the adult population, and therefore strictly correlated to cardiovascular events [22,23,24,25]. Childhood cardiac and vascular damage data are partial and very limited [26,27]. Identifying early cardiovascular dysfunctions during childhood, such as hypertension, cardiac remodeling or hypertrophy, arterial stiffness, and carotid intima-media thickness, could be important to prevent future cardiovascular events. Thus, the aim of this study is the evaluation of markers of early vascular damage and left ventricular geometry in a sample of children affected by ADPKD.
2. Materials and Methods
Patients with ADPKD were recruited from October to December 2018. These patients were referred to the Unit of Pediatric Nephrology of the University Hospital of Verona. The study was approved by the Ethical Committee of the University Hospital of Verona (CESC n.9427), and written informed consent was obtained from each participant’s parents.
Several vascular measurements were obtained. Specifically, ambulatory blood pressure monitoring (ABPM), office blood pressure (OBP), carotid intima-media thickness (cIMT), carotid distensibility (DC), pulse wave velocity (PWV), and echocardiographic measurements (relative wall thickness (RWT) and left ventricular mass index (LVMI)).
2.1. Blood Pressure Measurement and Vascular Exams
An oscillometric device recorded ABPM (Intermed A&D TM-2430). It was placed on the non-dominant arm and was set such that measurements were taken every 15 min during the day and every 30 min throughout the night, adapting “day” and “night” according to the diary form completed by the child or parents. All the values derived from the blood pressure (BP) measurements were also Z-score transformed according to normative values [28,29].
cIMT was assessed with an ultrasound of the carotid arteries (LogiQ P5 Pro, Bimedis, Kissimmee, FL, USA). The cIMT was estimated by tracking the artery wall in the last centimeter of the common carotid artery and calculated using dedicated hardware (Carotid studio, Quipu, Pisa, Italy). The relative z-scores and percentiles were calculated using reference values [30].
The cDC was calculated as cDC = ΔA/(A × ΔP), where A is the diastolic lumen area, ΔA is the stroke change in lumen area, and ΔP is pulse pressure (PP). Changes in diameters were detected using ultrasound B-mode image sequences of the right and left common carotid arteries acquired at different steps. These changes were analyzed using the above-mentioned automatic system [31]. The relative z-score and percentile were calculated according to reference values [32].
PWV was measured with SphygmoCor XCEL (AtCor Medical Pty Ltd.; Unit 11, West Ryde Corporate Centre, 1059–1063 Victoria Road, West Ryde, NSW 2114, Australia). To conduct a carotid–femoral PWV measurement, a cuff was placed around the femoral artery of the child to capture the femoral waveform, and a tonometer was used to capture the carotid waveform. The distance between the carotid and femoral arteries was measured, and the velocity was automatically determined by dividing the distance by the pulse transit time. The subtraction method calculated the distance between the carotid measurement site and the cuffed site. The distance was calculated from the sternal notch to the top edge of the femoral cuff (distal distance) and from the carotid artery to the sternal notch (proximal distance). To assess the above, the proximal distance was subtracted from the distal distance to determine the aortic lative z-score, and the percentile was calculated according to reference values [33].
A single sonographer performed transthoracic echocardiography (Esaote MyLabTM40, Roma, Italy) in all participants. In the parasternal long-axis view, the 2D method was used to measure interventricular septum thickness end-diastole (IVSd), left ventricular end-diastolic diameter (LVEDd), and left ventricular posterior wall thickness at end-diastole. The Relative Wall Thickness (RWT) was calculated through the following formula: (IVSd + LVPWd/LVEDd). The Devereux equation was used to obtain left ventricular mass (LVM = 0.80 * 1.04 [(tele-diastolic diameter + PW + IVS)3 − tele-diastolic diameter3] + 0.6 gr). LVM was indexed (LVMI) to height (m2.7) [34]. Left ventricular hypertrophy (LVH) was defined as the presence of an LVMI greater or equal to the 95th percentile, specific for age and sex [35]. The threshold for increased RWT (adjusted) was 0.375; the 95th percentile was specific for age [36]. Normal geometry was defined by normal LVMI and normal RWT, concentric remodeling by normal LVMI and increased RWT, concentric hypertrophy by increased LVMI and RWT, and eccentric hypertrophy by increased LVMI and normal RWT [37].
2.2. Laboratory Exams
Venous blood and urinary samples were collected after an overnight fast. Biochemical parameters such as triglycerides, total cholesterol, HDL cholesterol, LDL cholesterol, non-HDL cholesterol, uric acid, creatinine, cystatin C, microalbuminuria (spot urine sample), and proteinuria (24 h urine collection) were analyzed in a single reference centralized laboratory with standard methods on routine clinical chemistry instrumentation (Cobas 8000, Roche Diagnostics GmbH, Mannheim, Germany). The estimated glomerular filtration rate (eGFR) was calculated using the Schwartz equation.
2.3. Anthropometric Parameters
Anthropometric parameters were also assessed. Body weight was measured using a calibrated balance, and height was measured using a calibrated stadiometer. The body mass index (BMI) was calculated as weight (kg) divided by the square of height (m). Overweight and obesity were defined as BMI ≥ 85th and 95th percentile for sex and age, respectively [38]. The WHO reference for BMI categorizes children into the overweight and obese groups [39].
2.4. Statistical Analysis
For the statistical analysis, data were expressed as mean ± standard deviation for continuous variables or percentages for categorical ones. The Spearman correlation coefficient (rS) was used to quantify the linear relationship between variables. Statistical analyses were performed using SPSS software (IBM Corp. Released 2015. IBM SPSS Statistics for Windows, Version 23.0. Armonk, NY, USA: IBM Corp). Graphs were created with GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla, CA, USA (www.graphpad.com).
3. Results
Patients’ characteristics are reported in .
Eleven patients, seven males and four females, were included in the study. The mean age of the population was 9.5 ± 3.2 years. The median BMI was 18.9 ± 3.4 kg/m2. Three children were overweight (>85th), and eight were normal weight. None of the subjects had altered glomerular filtration rate (GFR), and the mean GFR was 109.6 ± 13.5 mL/min/m2. The mean microalbuminuria measured by the albumin mg/mmol ratio was 1.1 ± 1.3. The cardiovascular parameters according to the percentile reference values are reported in .
Two ABPM, one cIMT, three cDC, and two PWV measurements were unavailable. Four children had high blood pressure (HBP) according to ABPM, five were normotensive, and only two patients were hypertensive at office blood pressure (OBP) measurement, consistent with the possible diagnosis of masked hypertension. One child was already under chronic therapy with an ACE inhibitor. RWT was, on average, higher concerning the cut-off for remodeling (mean 0.39 ± 0.05). In particular, eight patients had concentric cardiac remodeling, while one had cardiac concentric hypertrophy. cIMT was above the 95th percentile for sex and height in 80% of children (0.5 ± 0.005 mm), while average PWV and cDC were within the normal range (5.5 ± 4.6 m/s and 89.6 ± 16.1 × 10−3/KPa, respectively). We observed a positive correlation between the PWV and RWT (rS = 0.616; p = 0.044; Figure 1a) and a negative correlation between cDC and RWT (rS = −0.770; p = 0.015; Figure 1b), suggesting a close relationship between vascular dysfunction and initial cardiac damage.
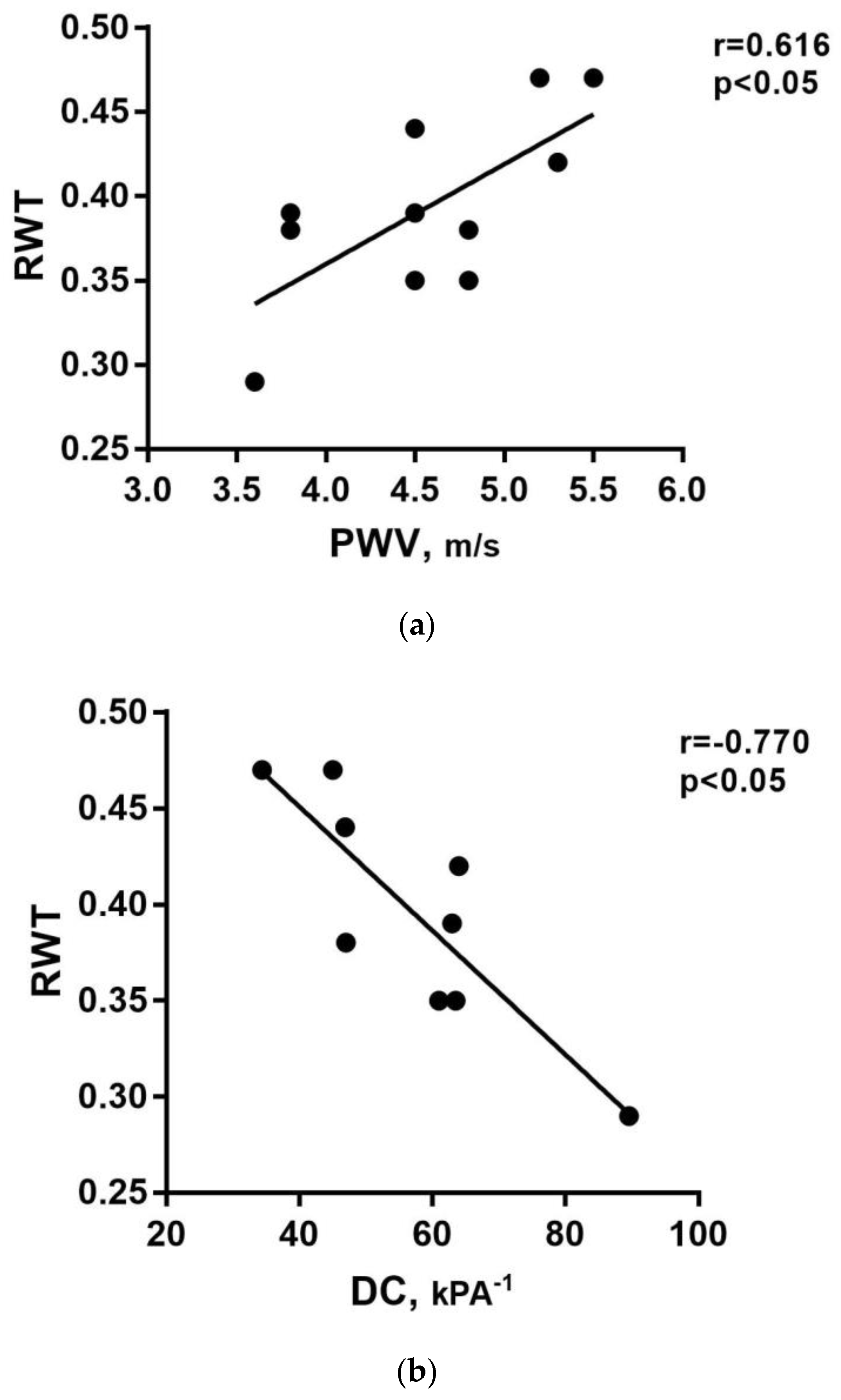
Figure 1. (a) Linear positive correlation between the pulse wave velocity (PWV) and relative wall thickness (RWT). (b) Linear negative correlation between carotid distensibility coefficient (cDC) and RWT.
Of note, cardiovascular damages were also found in normotensive patients; three patients already had cardiac remodeling, one had cardiac concentric hypertrophy, and four were in the IMT ≥ 95° percentile. Probably, because of the early age of our population, we found more concentric remodeling than cardiac hypertrophy. No correlations between cardiovascular parameters and either proteinuria or GFR were found.
4. Discussion
This study showed the prevalence of early organ damage in children affected by ADPKD. Nowak et al. [26] demonstrated vascular dysfunction measured by brachial artery flow-mediated dilation and arterial stiffness, measured as carotid–femoral pulse wave velocity. Still, it was measured in young adults with a mean age of 21. In addition, they did not assess cardiac parameters. In the only other paper conducted on children, Karava et al. [27] demonstrated in 21 adolescents a high PWV and increased cIMT in comparison with matched controls, indicating an increase in arterial stiffness and hypertrophic vasculopathy. They also found that around 10% of patients had left ventricular hypertrophy (but only in patients on antihypertensive treatment) and a linear correlation between LVH and PWV and LVH and cIMT. Patients with LVH were older, suggesting that arterial dysfunction precedes cardiac damage. Cardiac remodeling RWT was not assessed. A total of 19% of patients had hypertension. Of note, even Karava’s population was older than ours because the mean age was 12 years. Few other studies on adolescents with ADPKD showed a higher LVMI in hypertensive and borderline hypertensive adolescents than non-hypertensive [40]. A higher LVMI in patients with ADPKD as compared to group control was also shown, although within the normal range [41,42]. It has also been reported that around half of adult patients with ADPKD have LVH [43]. All the remaining studies investigating cardiac and vascular damage in patients with ADPKD were conducted in adults [14,15,17,44,45,46]. In the present study, we have shown a very early vascular and cardiac impairment in a patient affected by ADPKD despite a mean age of only 9.5 years. We have found neither PWV nor DC beyond the reference range, but different from the study of Karava et al. [27], we showed that 80% of patients had a cIMT higher than the 95th percentile. Regarding cardiac damage, the mean RWT was increased on average, and this was a sign of cardiac remodeling, whereas only one child had cardiac concentric hypertrophy. This is probably due to the very young age of our population, with concentric remodeling being a (possible) first step before overt cardiac hypertrophy can develop. It is also intuitive that vascular dysfunction precedes cardiac damage. Finally, we found a positive correlation between PWV and RWT, as well as a negative correlation between DC and RWT. Taken together, these unexpected findings suggest that vascular and cardiac damage is already detectable in children <10 years and that they are in part related to each other. This evidence is in line with the fact that most ADPKD patients die because of cardiovascular diseases [19,47]. Despite the evidence of cardiac and vascular damage, only two patients out of eleven in our sample could be classified as hypertensive if evaluated using office BP. Conversely, if we refer to ABPM, the number of hypertensive children rises to four out of ten, compatible with a diagnosis of masked hypertension. This suggests that hypertension is underestimated, and a widespread use of ABPM should probably be advised as a better screening tool in all patients affected by ADPKD. In previous studies using office BP, the median age for hypertension diagnosis in patients with ADPKD was 33 years in males and 38 years in females whose parents were hypertensive and 40 years in males and 50 years in females whose parents were non-hypertensive [48]. Interestingly, in our sample, target organ damage was not found only in hypertensive children but also in some normotensive ones. This is a significant finding, suggesting that cardiovascular injuries in these patients are not only related to hypertension. This hypothesis is consistent with the fact that the damage caused by hypertension takes several years. Due to the young age of our population, one can speculate that other mechanisms can subtend cardiovascular damage besides and beyond hypertension per se.
The reason why patients with ADPKD develop so early and so frequently experience cardiovascular damage is still debated. The activation of the renin-angiotensin-aldosterone-system (RAAS) [17,18,49,50,51,52], an increased sympathetic tone [53], and endothelial dysfunction, primarily via endothelin and nitric oxide actions [16,54,55,56,57,58,59], were intensively investigated.
According to the results of our study, the role of blood pressure/hypertension is not pivotal since the markers of early vascular and cardiac damage are not different in children with or without high blood pressure , and cardiovascular damage was present in normotensive patients.
Anyhow, it is conceivable to speculate that early antihypertensive treatment, especially with drugs that modulate renin-angiotensin-aldosterone, may be important for preventing renal worsening or cardiovascular protection. Both these speculations are supported by the notion that cardiovascular disease prevention occurs by reducing blood pressure and LVH, even in ADPKD patients [60,61]. So far, the use of ACE inhibitors and maintaining blood pressure < 120/80 mmHg are recommended in hypertensive patients with ADPKD [62]. By contrast, if ACE inhibitors have been shown to slow kidney disease progression in patients with nephropathies, especially if proteinuria is present, the role of ACE inhibitors in slowing the progression of ADPKD needs to be clarified. In a randomized, prospective trial, after a 5-year follow-up in patients with ADPKD who had well-preserved renal function, amlodipine and enalapril were associated with a similar decline in creatinine clearance. Still, only enalapril showed a sustained antialbuminuric effect leading to higher protective effects in the long term [63]. No differences in the loss of renal function were also found in comparison with beta-blockers after a 3-year follow-up [64]. On the other hand, it has been demonstrated that lowering blood pressure is crucial in slowing renal damage progression regardless of antihypertensive use [64,65]. Even a meta-analysis including 142 patients affected by ADPKD concluded that ACE inhibitors are more effective in lowering urine protein excretion. Still, the progression rate of kidney disease was not significantly slower than other agents [62]. All these data are in line with the hypothesis that, for slowing kidney disease progression in ADPKD, lowering blood pressure is particularly important, regardless of the therapy used, even though a more extended trial may be needed.
The cross-sectional design of the present study does not allow us to see if cardiac and vascular damages develop contemporarily or are, in part, causes or consequences of each other. Another limitation of the present study is that our population consisted of only 11 patients. However, the rarity of the disease can, at least partially, justify the restricted number of included patients, which is consistent with other studies [26,27,41]. We also have to acknowledge the lack of a control group. Reference values were taken from previous epidemiological studies on children from European or US populations [32,33,34,35,36]. Moreover, another limit was that genetics data were not available.
5. Conclusions
In conclusion, in this study, we showed a high prevalence of organ damage even in children (with an average age < 10 years old) affected by ADPKD. In these children, we report tendentially increased RWT and high cIMT, with likely masked hypertension in almost half of patients despite normal renal function. Although this observation is clinically relevant, the present study should be read cautiously due to several limitations. First of all, because of the small sample size, this remains a pilot study, and firm conclusions cannot be drawn. Second, a control group is missing, and thus differences between ADPKD patients and healthy children cannot be determined, but only interpreted in the light of references values. Anyhow, our data should be a warning and prompt towards early cardiovascular screening and BP measurements using ABPM in children affected by ADPKD. Future studies using a higher sample size and adequate control groups may help to address the unmet points raised by our pilot study.
References
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Srivastava, A.; Patel, N. Autosomal dominant Polycystic kidney disease. Am. Fam. Physician 2014, 90, 303–307. [Google Scholar]
- Harris, P.C.; Torres, V.E. Polycystic Kidney Disease, Autosomal Dominant; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a Gene for Polycystic Kidney Disease That Encodes an Integral Membrane Protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef]
- Huang, E.; Samaniego-Picota, M.; McCune, T.; Melancon, J.K.; Montgomery, R.A.; Ugarte, R.; Kraus, E.; Womer, K.; Rabb, H.; Watnick, T. DNA Testing for Live Kidney Donors at Risk for Autosomal Dominant Polycystic Kidney Disease. Transplantation 2009, 87, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Gabow, P.A.; Duley, I.; Johnson, A.M. Clinical Profiles of Gross Hematuria in Autosomal Dominant Polycystic Kidney Disease. Am. J. Kidney Dis. 1992, 20, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Johnson, A.M.; Gabow, P.A.; Schrier, R.W. Overt proteinuria and microalbuminuria in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1994, 5, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Nishiura, J.A.A.L.; Neves, R.F.; Eloi, S.R.; Cintra, S.M.; Ajzen, S.A.; Heilberg, I.P. Evaluation of Nephrolithiasis in Autosomal Dominant Polycystic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 838–844. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Audrézet, M.P.; Rousseau, A.; Hourmant, M.; Renaudineau, E.; Charasse, C.; Morin, M.-P.; Moal, M.-C.; Dantal, J.; Wehbe, B.; et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 942–951. [Google Scholar] [CrossRef]
- Chapman, A.B.; Rubinstein, D.; Hughes, R.; Stears, J.C.; Earnest, M.P.; Johnson, A.M.; Gabow, P.A.; Kaehny, W.D. Intracranial Aneurysms in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 1992, 327, 916–920. [Google Scholar] [CrossRef]
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Manco-Johnson, M.L.; Duley, I.T.; Everson, G.T. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 1990, 11, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Blumenfeld, J.D.; Chhabra, S.; Dutruel, S.P.; Thimmappa, N.D.; Bobb, W.O.; Donahue, S.; Rennert, H.E.; Tan, A.Y.; Giambrone, A.E.; et al. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology 2016, 280, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Hossack, K.F.; Leddy, C.L.; Johnson, A.M.; Schrier, R.W.; Gabow, P.A. Echocardiographic Findings in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 1988, 319, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Massella, L.; Mekahli, D.; Paripović, D.; Prikhodina, L.; Godefroid, N.; Niemirska, A.; Ağbaş, A.; Kalicka, K.; Jankauskiene, A.; Mizerska-Wasiak, M.; et al. Prevalence of Hypertension in Children with Early-Stage ADPKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 874–883. [Google Scholar] [CrossRef]
- Griffin, M.D.; Torres, V.E.; Grande, J.P.; Kumar, R. Vascular expression of polycystin. J. Am. Soc. Nephrol. 1997, 8, 616–626. [Google Scholar] [CrossRef]
- Chapman, A.B.; Stepniakowski, K.; Rahbari-Oskoui, F. Hypertension in Autosomal Dominant Polycystic Kidney Disease. Adv. Chronic Kidney Dis. 2010, 17, 153–163. [Google Scholar] [CrossRef]
- Torres, V.E.; Donovan, K.A.; Scicli, G.; Holley, K.E.; Thibodeau, S.N.; Carretero, O.A.; Inagami, T.; McAteer, J.A.; Johnson, C.M. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int. 1992, 42, 364–373. [Google Scholar] [CrossRef]
- Fick, G.M.; Johnson, A.M.; Hammond, W.S.; Gabow, P.A. Causes of death in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1995, 5, 2048–2056. [Google Scholar] [CrossRef]
- Perrone, R.D.; Ruthazer, R.; Terrin, N.C. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: Contribution of extrarenal complications to mortality. Am. J. Kidney Dis. 2001, 38, 777–784. [Google Scholar] [CrossRef]
- Rahman, E.; Niaz, F.A.; Al-Suwaida, A.; Nahrir, S.; Bashir, M.; Rahman, H.; Hammad, D. Analysis of causes of mortality in patients with autosomal dominant polycystic kidney disease: A single center study. Saudi J. Kidney Dis. Transplant. 2009, 20, 806–810. [Google Scholar]
- Chung, C.-M.; Lin, Y.-S.; Chang, S.-T.; Cheng, H.-W.; Yang, T.-Y.; Hsiao, J.-F.; Pan, K.-L.; Hsu, J.-T.; Chu, C.-M. Arterial Stiffness Is the Independent Factor of Left Ventricular Hypertrophy Determined by Electrocardiogram. Am. J. Med. Sci. 2012, 344, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Lekakis, J.; Papadopoulos, C.; Triantafyllidi, H.; Paraskevaidis, I.; Georgoula, G.; Tzortzis, S.; Revela, I.; Kremastinos, D.T. Incremental Value of Pulse Wave Velocity in the Determination of Coronary Microcirculatory Dysfunction in Never-treated Patients with Essential Hypertension. Am. J. Hypertens. 2008, 21, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.W.; Markus, H.S.; Bots, M.L.; Rosvall, M.; Sitzer, M. Prediction of Clinical Cardiovascular Events with Carotid Intima-Media Thickness. Circulation 2007, 115, 459–467. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Aznaouridis, K.; Stefanadis, C. Prediction of Cardiovascular Events and All-Cause Mortality with Arterial Stiffness. J. Am. Coll. Cardiol. 2010, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Farmer, H.; Cadnapaphornchai, M.A.; Gitomer, B.; Chonchol, M. Vascular dysfunction in children and young adults with autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2017, 32, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Karava, V.; Benzouid, C.; Hogan, J.; Dossier, C.; Denjean, A.P.; Deschênes, G. Early cardiovascular manifestations in children and adolescents with autosomal dominant polycystic kidney disease: A single center study. Pediatr. Nephrol. 2018, 33, 1513–1521. [Google Scholar] [CrossRef]
- Wühl, E.; Witte, K.; Soergel, M.; Mehls, O.; Schaefer, F.; German Working Group on Pediatric Hypertension. Distribution of 24-h ambulatory blood pressure in children: Normalized reference values and role of body dimensions. J. Hypertens. 2002, 20, 1995–2007. [Google Scholar] [CrossRef] [PubMed]
- National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 2004, 114 (Suppl. S2), 555–576. [Google Scholar] [CrossRef]
- Pozza, R.D.; Ehringer-Schetitska, D.; Fritsch, P.; Jokinen, E.; Petropoulos, A.; Oberhoffer, R. Intima media thickness measurement in children: A statement from the Association for European Paediatric Cardiology (AEPC) Working Group on Cardiovascular Prevention endorsed by the Association for European Paediatric Cardiology. Atherosclerosis 2015, 238, 380–387. [Google Scholar] [CrossRef]
- Bianchini, E.; Bozec, E.; Gemignani, V.; Faita, F.; Giannarelli, C.; Ghiadoni, L.; Demi, M.; Boutouyrie, P.; Laurent, S. Assessment of carotid stiffness and intima-media thickness from ultrasound data: Comparison between two methods. J. Ultrasound Med. 2010, 29, 1169–1175. [Google Scholar] [CrossRef]
- Doyon, A.; Kracht, D.; Bayazit, A.K.; Deveci, M.; Duzova, A.; Krmar, R.T.; Litwin, M.; Niemirska, A.; Oguz, B.; Schmidt, B.M.W.; et al. Carotid artery intima-media thickness and distensibility in children and adolescents: Reference values and role of body dimensions. Hypertension 2013, 62, 550–556. [Google Scholar] [CrossRef]
- Elmenhorst, J.; Hulpke-Wette, M.; Barta, C.; Pozza, R.D.; Springer, S.; Oberhoffer, R. Percentiles for central blood pressure and pulse wave velocity in children and adolescents recorded with an oscillometric device. Atherosclerosis 2015, 238, 9–16. [Google Scholar] [CrossRef]
- De Simone, G.; Devereux, R.B.; Daniels, S.R.; Koren, M.J.; Meyer, R.A.; Laragh, J.H. Effect of growth on variability of left ventricular mass: Assessment of allometric signals in adults and children and their capacity to predict cardiovascular risk. J. Am. Coll. Cardiol. 1995, 25, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Khoury, P.R.; Mitsnefes, M.; Daniels, S.R.; Kimball, T.R. Age-Specific Reference Intervals for Indexed Left Ventricular Mass in Children. J. Am. Soc. Echocardiogr. 2009, 22, 709–714. [Google Scholar] [CrossRef]
- De Simone, G.; Daniels, S.R.; Kimball, T.R.; Roman, M.J.; Romano, C.; Chinali, M.; Galderisi, M.; Devereux, R.B. Evaluation of concentric left ventricular geometry in humans: Evidence for age-related systematic underestimation. Hypertension 2005, 45, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Ganau, A.; Devereux, R.B.; Roman, M.J.; de Simone, G.; Pickering, T.G.; Saba, P.S.; Vargiu, P.; Simongini, I.; Laragh, J.H. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J. Am. Coll. Cardiol. 1992, 19, 1550–1558. [Google Scholar] [CrossRef]
- Barlow, S.E.; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: Summary report. Pediatrics 2007, 120 (Suppl. S4), S164–S192. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Bellizzi, M.C.; Flegal, K.M.; Dietz, W.H. Establishing a standard definition for child overweight and obesity worldwide: International survey. BMJ 2000, 320, 1240. [Google Scholar] [CrossRef]
- Cadnapaphornchai, M.A.; McFann, K.; Strain, J.D.; Masoumi, A.; Schrier, R.W. Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int. 2008, 74, 1192–1196. [Google Scholar] [CrossRef]
- Zeier, M.; Geberth, S.; Schmidt, K.G.; Mandelbaum, A.; Ritz, E. Elevated blood pressure profile and left ventricular mass in children and young adults with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1993, 3, 1451–1457. [Google Scholar] [CrossRef]
- Saggar-Malik, A.K.; Missouris, C.G.; Gill, J.S.; Singer, D.R.J.; Markandu, N.D.; MacGregor, G.A. Left ventricular mass in normotensive subjects with autosomal dominant polycystic kidney disease. BMJ 1994, 309, 1617–1618. [Google Scholar] [CrossRef]
- Chapman, A.B.; Johnson, A.M.; Rainguet, S.; Hossack, K.; Gabow, P.; Schrier, R.W. Left ventricular hypertrophy in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1997, 8, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Jin, X.; Ye, C.; Chen, J.; Mei, C. Carotid vascular remodelling in patients with autosomal dominant polycystic kidney disease. Nephrology 2009, 14, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, K.; Oflaz, H.; Uslu, B.; Cimen, A.O.; Elitok, A.; Kasikcioglu, E.; Alisir, S.; Tufan, F.; Namli, S.; Uysal, M.; et al. Coronary Flow Velocity Reserve and Carotid Intima Media Thickness in Patients with Autosomal Dominant Polycystic Kidney Disease: From Impaired Tubules to Impaired Carotid and Coronary Arteries. Clin. J. Am. Soc. Nephrol. 2008, 3, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Kocaman, O.; Oflaz, H.; Yekeler, E.; Dursun, M.; Erdogan, D.; Demirel, S.; Alisir, S.; Turgut, F.; Mercanoglu, F.; Ecder, T. Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 2004, 43, 854–860. [Google Scholar] [CrossRef]
- Ecder, T.; Schrier, R.W. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat. Rev. Nephrol. 2009, 5, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Johnson, A.M.; Mcfann, K.; Chapman, A.B. The role of parental hypertension in the frequency and age of diagnosis of hypertension in offspring with autosomal-dominant polycystic kidney disease. Kidney Int. 2003, 64, 1792–1799. [Google Scholar] [CrossRef]
- Gabow, P.A.; Chapman, A.B.; Johnson, A.M.; Tangel, D.J.; Duley, I.T.; Kaehny, W.D.; Manco-Johnson, M.; Schrier, R.W. Renal structure and hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1990, 38, 1177–1180. [Google Scholar] [CrossRef]
- Fick, G.M.; Duley, I.T.; Johnson, A.M.; Strain, J.D.; Manco-Johnson, M.L.; Gabow, P.A. The spectrum of autosomal dominant polycystic kidney disease in children. J. Am. Soc. Nephrol. 1994, 4, 1654–1660. [Google Scholar] [CrossRef]
- Chapman, A.B.; Johnson, A.; Gabow, P.A.; Schrier, R.W. The Renin–Angiotensin–Aldosterone System and Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 1990, 323, 1091–1096. [Google Scholar] [CrossRef]
- Harrap, S.B.; Davies, D.L.; Macnicol, A.M.; Dominiczak, A.F.; Fraser, R.; Wright, A.F.; Watson, M.L.; Briggs, J.D. Renal, cardiovascular and hormonal characteristics of young adults with autosomal dominant polycystic kidney disease. Kidney Int. 1991, 40, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.H.H.T.; Ligtenberg, G.; Oey, P.L.; Koomans, H.A.; Blankestijn, P.J. Sympathetic Activity Is Increased in Polycystic Kidney Disease and Is Associated with Hypertension. J. Am. Soc. Nephrol. 2001, 12, 2427–2433. [Google Scholar] [CrossRef] [PubMed]
- Giusti, R.; Neri, M.; Angelini, D.; Carlini, A.; Fiorini, I.; Bigongiari, P.; Antonelli, A. Plasma Concentration of Endothelin and Arterial Pressure in Patients with ADPKD. In Autosomal Dominant Polycystic Kidney Disease; Karger Publishers: Basel, Switzerland, 1995; Volume 115, pp. 118–121. [Google Scholar] [CrossRef]
- Hocher, B.; Zart, R.; Schwarz, A.; Vogt, V.; Braun, C.; Thöne-Reineke, C.; Braun, N.; Neumayer, H.H.; Koppenhagen, K.; Bauer, C.; et al. Renal endothelin system in polycystic kidney disease. J. Am. Soc. Nephrol. 1998, 9, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Munemura, C.; Uemasu, J.; Kawasaki, H. Epidermal Growth Factor and Endothelin in Cyst Fluid from Autosomal Dominant Polycystic Kidney Disease Cases: Possible Evidence of Heterogeneity in Cystogenesis. Am. J. Kidney Dis. 1994, 24, 561–568. [Google Scholar] [CrossRef]
- Wang, D.; Iversen, J.; Strandgaard, S. Endothelium-Dependent Relaxation of Small Resistance Vessels Is Impaired in Patients with Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2000, 11, 1371–1376. [Google Scholar] [CrossRef]
- Wang, D.; Iversen, J.; Wilcox, C.S.; Strandgaard, S. Endothelial dysfunction and reduced nitric oxide in resistance arteries in autosomal-dominant polycystic kidney disease. Kidney Int. 2003, 64, 1381–1388. [Google Scholar] [CrossRef]
- Torres, V.E.; Cai, Y.; Chen, X.; Wu, G.Q.; Geng, L.; Cleghorn, K.A.; Johnson, C.M.; Somlo, S. Vascular Expression of Polycystin-2. J. Am. Soc. Nephrol. 2001, 12, 1–9. [Google Scholar] [CrossRef]
- Ecder, T. Reversal of left ventricular hypertrophy with angiotensin converting enzyme inhibition in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 1999, 14, 1113–1116. [Google Scholar] [CrossRef]
- Schrier, R.; McFann, K.; Johnson, A.; Chapman, A.; Edelstein, C.; Brosnahan, G.; Ecder, T.; Tison, L. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: Results of a seven-year prospective randomized study. J. Am. Soc. Nephrol. 2002, 13, 1733–1739. [Google Scholar] [CrossRef]
- Jafar, T.H.; Stark, P.C.; Schmid, C.H.; Strandgaard, S.; Kamper, A.-L.; Maschio, G.; Becker, G.; Perrone, R.D.; Levey, A.S.; ACE Inhibition in Progressive Renal Disease (AIPRD) Study Group. The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int. 2005, 67, 265–271. [Google Scholar] [CrossRef]
- Ecder, T.; Chapman, A.B.; Brosnahan, G.M.; Edelstein, C.L.; Johnson, A.M.; Schrier, R.W. Effect of antihypertensive therapy on renal function and urinary albumin excretion in hypertensive patients with autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 2000, 35, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.A.; Breuning, M.H.; Duiser, R.; van Es, L.A.; Westendorp, R.G.J. No effect of enalapril on progression in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2003, 18, 2314–2320. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, R.; Poliak, R.; Stiasny, B.; Schmieder, R.E.; Schulze, B.D. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2007, 23, 573–579. [Google Scholar] [CrossRef] [PubMed]