


1. Introduction
Poor aqueous solubility is a major concern for new chemical entities (NCEs) and many of the existing active pharmaceutical ingredients (APIs) developed for oral drug delivery, where these substances have great pharmacological and permeability potential. An increasing proportion of potential new drug candidates (40–70%) are classified as Biopharmaceutical Classification System (BCS) Class II or IV drugs, and the formulation and commercialisation of these as oral dosage forms presents great challenges as they show sub-optimal dissolution; absorption; and, hence, bioavailability [1]. The poor aqueous solubility has a negative effect on dissolution, resulting in the incomplete absorption of these APIs in the gastrointestinal tract; hence, the bioavailability is affected detrimentally [2,3]. Several techniques are used to overcome this problem, which include physical and chemical modifications of the drug through micronization, crystal engineering, salt formation, the addition of surfactants, solid dispersions with hydrophilic carriers, complexation, hydrotropy, eutectic mixtures, and amorphous systems, to mention but just a few [2,4]. Among these solid-state alterations, solid dispersions and co-amorphous and co-crystal systems have taken the lead over the past two decades. One of the common factors among these techniques that plays a significant role in solubility and bioavailability enhancement is the type of carrier, co-former, or excipient used [5,6]. In the selection of a carrier, preference is given to carriers that are hydrophilic in nature [5], have high hydrogen bonding propensity, have higher glass transition temperatures (Tg), are non-hygroscopic [7], and form part of the United States Food and Drug Administration (FDA)-approved Generally Recognized as Safe (GRAS) list [8].
Naturally occurring sugars of several types, such as monosaccharides (glucose, fructose, galactose); disaccharides (sucrose, maltose, lactose); and sugar alcohols (polyols) such as sorbitol, mannitol, xylitol, erythritol, maltitol, lactitol, and isomalt, have been reported to enhance the solubility of drugs. The FDA designates sugars and polyols as GRAS excipients [9,10], and in addition to their proven safety, these compounds exhibit good water solubility [11] and possess hydroxyl groups (-OH) attached to each carbon atom in the molecule, which, in theory, provides hydrogen-bonding sites for drug molecules, thereby already meeting three of the abovementioned criteria in terms of suitable solid-state co-formers.
There are several ways in which sugars and polyols have been used to enhance the solubility of drugs. One predominant method used is to form solid dispersions of the drug [12]; another approach is the formulation of eutectic mixtures [13,14]. Polyols such as xylitol [15,16], mannitol [15], and sorbitol [17] have been reported as potential co-formers for developing co-crystalline systems. However, efforts made in exploring sugars and polyols in the making of co-crystals and co-amorphous systems are few to none. The low molecular weight, safety, good glass-forming ability, and (in some instances) high Tg of sugars [18] make them an interesting option to explore as excipients for drug–excipient co-amorphous and co-crystalline systems. The current review focuses on summarising the use of naturally occurring sugars and polyols for the enhancement of solubility and the dissolution rate of poorly soluble drugs. Furthermore, it seeks to understand the pros and cons in relation to the various techniques used; to provide a theoretical basis for their application in the formulation of co-amorphous and co-crystalline systems; and finally, to identify potential opportunities for future research.
2. Solid Dispersions
Since first introduced by Sekiguchi and Obi in 1961, solid dispersions (SDs) have been the most predominantly used solid-state modification technique, found to have significant commercial success [19,20,21]. Since the first FDA-approved SD, Cesamet® in 1985 (Bausch Health Companies Inc., Laval, QB, Canada), over 30 amorphous solid dispersions of Class II and Class IV drugs are now currently marketed as oral dosage forms, signifying the importance of this technique [20,21,22]. A solid dispersion (SD) can be defined as a solid-state mixture of one or more hydrophobic drugs and one or more hydrophilic carriers. The process involves the distribution of the drug molecules in the hydrophilic carrier matrix at the molecular or colloidal level via melting (fusion), quench cooling, solvent evaporation, spray drying, freeze drying, or hot-melt extrusion [5,23]. In SDs, either the particle size of the drug is reduced, resulting in the increased surface area of the drug upon contact with the solvent, or a crystalline pure drug is converted into an amorphous form leading to increased solubility [23]. Based on the miscibility of the drug in the carrier, the final solid-state form (crystalline or amorphous), and the molecular arrangement, SDs can be classified as (a) simple eutectic mixtures, (b) solid solutions, (c) glass solutions, (d) glass suspensions, or (e) amorphous precipitations in a crystalline carrier [5,23,24]. A summary of different types of SDs and their solid states is illustrated in Figure 1.
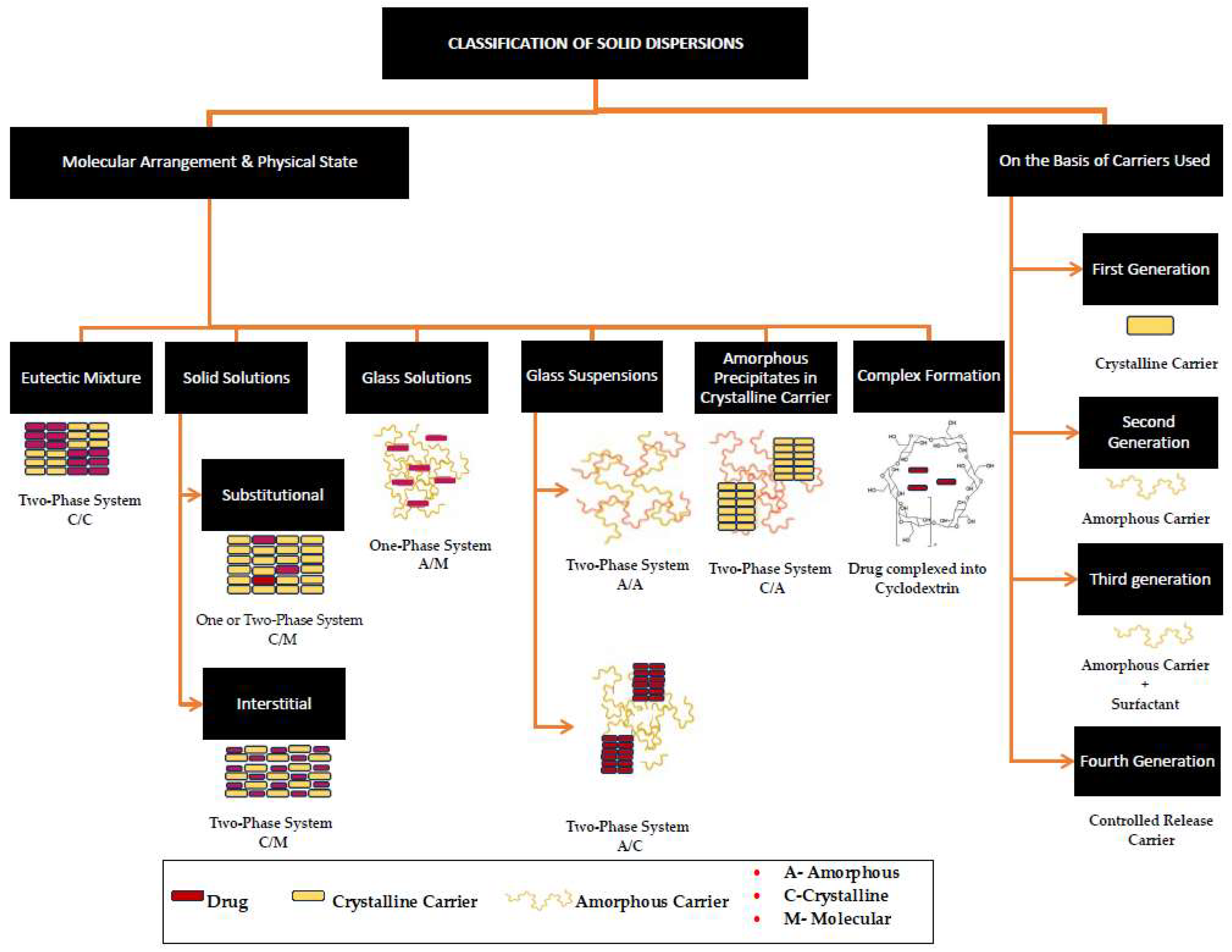
Figure 1. A schematic depiction of the classification of solid dispersions, obtained and reproduced with changes from Ref. [25] and the Association of Pharmaceutical Teachers of India in accordance with Creative Commons Attribution—Non-Commercial 4.0 International (CC BY-NC 4.0).
SDs carry common properties in the sense that the carriers used are typically hydrophilic and inert and exhibit the common principles of solubility and dissolution enhancement. The main and common mechanisms of solubility enhancement are a reduction in the particle size of the crystallite API to its molecular size for molecular dispersion or ultrafine particulates in the making of solid suspension. This increase in surface area results in the enhanced wettability and dispersibility of the drug into the dissolution medium [5,26]. According to the BCS, Class II drugs exhibit a dissolution rate of limited absorption, and therefore, solid dispersion technology is a promising approach to enhancing dissolution. While the commercial success of BCS Class II drugs is limited by their inherent low dissolution, Class IV drugs show permeation-limited absorption. Among the various physical and chemical modification techniques available, SDs provide a wide range of opportunities, as they offer greater flexibility in formulating oral drug delivery systems given the availability of various processing and excipient alternatives [19]. The ease of preparation and regulatory compliance requirements of SDs in comparison with techniques such as those involving the formation of solvates, hydrates, salt formation, the inclusion of polar or ionizable groups, and co-crystallisation makes them a more favourable approach [22,27,28].
SDs can be prepared by applying a wide range of techniques, including simple fusion; hot-melt extrusion that uses heating; solvent-based preparation techniques such as simple kneading with selected solvents; solvent evaporation; spray-drying; freeze-drying; and solvent-free methods such as milling with a ball-mill or air-jet milling. Though the preparation of SDs is relatively simple, they are accompanied by unique challenges such as the thermal instability of their APIs and carriers; a lack of solubility in the hydrophilic carriers in organic solvents, leading to phase separation during the process; high costs; low yields associated with techniques such as spray-drying; and often, scalability for commercial purposes. A summary of various techniques used in the preparation of SDs, their advantages and limitations are discussed in the following table .
2.1. Solid Dispersions of Sugar Carriers
A variety of hydrophilic carriers have been reported in the formulation of SDs, wherein crystalline carriers such as sugars have been studied in the formulation of first-generation Class C-C (crystalline drug dispersed in crystalline carrier) SDs [23,34].
Allen et al. [35] first reported the use of sugars in the preparation of SDs for the solubility enhancement of several corticosteroids. Sugar-based glass dispersions were prepared using the fusion method, where the sugars (dextrose, galactose, and sucrose) formed an amorphous environment for the dispersion of the drug molecules when rapidly cooled from a molten state. The authors reported a good glass-forming ability and an increased dissolution rate for the drug for all the sugars used. Dissolution studies indicated a bi-phasic drug release with rapid initial drug release followed by prolonged drug release. This phenomenon was attributed to the formation of partial glass solutions in the sugars, as they were not heated to the melting point to avoid the degradation of the corticosteroids. This resulted in the partial distribution of the drug at the molecular level. Despite this, SDs were found to increase the dissolution rate of the drugs in comparison with the pure, unaltered drug. The improved dissolution rate was due to a reduction in the particle size of the drug to a very fine state (Phase—I) and increased wettability (Phase—II). The study also reported some challenges such as discolouration, especially observed for sucrose and dextrose, indicating the degradation of the sugars and the hygroscopic nature of the resulting SDs.
To overcome these challenges, Allen et al. [36] prepared ternary sugar-based SD systems of corticosteroids with sucrose–mannitol (1:1) and sorbitol–mannitol (1:1) in a drug-to-carrier ratio of 1:19. The results were indicative of the good miscibility of the drugs in the sugar carrier systems, less hygroscopicity, and no discolouration. This was mainly attributable to the presence of mannitol in the mixtures, as the binary solid dispersion of mannitol as a sugar carrier formed a stable, non-hygroscopic, free-flowing dispersion system that revealed no discolouration during melting and good miscibility with the drugs. Interestingly, the lack of discolouration in the sucrose–mannitol systems was due to a reduction in the melting point of the system, indicating the formation of a eutectic mixture. Tablets prepared from these solid dispersions showed a significant increase in the dissolution rate over the tablets with the drug alone (30% vs. 80% drug release in 10 min). This was ascribed to the enhanced wettability of the drug particles facilitated by the hydrophilic sugar carriers.
Ghanem et al. [37] prepared SDs made of sulfamethoxazole via the quench-cooling technique using non-reducing sugars (glucose and galactose) and reducing sugars (maltose and sucrose). SDs consisting of a drug-to-sugar ratio of 1:1 were prepared by heating the mixture slightly above the melting point of the individual sugars to avoid caramelisation, which resulted in a glassy material with sulfamethoxazole dispersed as solid particles. In comparison with the drug, which showed only approximately 20% drug dissolution, the SDs consisting of glucose and maltose showed 100% drug dissolution within 5 min, while the galactose-based SDs showed complete drug dissolution only after 90 min. Similar results were reported in earlier studies [35], where drug releases from galactose-based SDs were low in comparison with other sugars and at times lower than the drug itself. Similar to previous studies, the increase in the solubility of the solid dispersions was attributable to the wettability of the drug particles.
The authors further reported forming a complex between the free carbonyl group of the non-reducing sugars and the amino group of the drug. This complexation could have potentially resulted in a new solid-sate form of the APIs, which is worth further investigation using advanced characterisation techniques such as differential scanning calorimetry (DSC) and powder X-ray diffraction (PXRD). It was also reported that the complexation increased with an increase in the weight ratio of the sugar, as high as 1:50 for full complexation in the case of glucose. The study also reported that the observed complexation only occurred after using the fusion technique, whereas solvent evaporation from a drug:sugar mixture dissolved in an organic solvent, such as methanol, did not result in drug:sugar complexation. This is owing to the poor solubility of sugars in organic solvents [38] and the fact that the use of a common solvent does not necessarily have a positive effect on the chemical interaction between a drug and a carrier [8].
SDs of carbamazepine (CBZ) and nitrazepam were prepared using lactose and galactose via the fusion method at room temperature. The resulting sugar-based SD-system, consisting of a CBZ-to-carrier ratio of 1:3, showed an enhanced dissolution rate with a bi-phasic drug release profile, similar to the studies discussed above. In another study, SDs of CBZ were prepared via a quench-cooling process using lactose as the carrier. The results indicated an increase in the dissolution rate with an increase in the carrier concentration. The enhanced dissolution was mainly attributable to the reduced particle size and increased wettability of the drug, increasing the chances of hydrogen bond formation between the hydroxyl group of lactose and the carboxylic acid group of the CBZ [39]. In addition to the findings mentioned above, the authors also noted that the dissolution rates of the SDs were more consistent than those of the physical mixtures and coprecipitates. Both these studies confirm the partial amorphization of the drug, which is the main reason for an enhanced dissolution rate.
In preparing ibuprofen SDs via the fusion method, sugar carriers (icing sugar (sucrose), dextrose, mannitol, and lactose) have been used and compared against the dissolution rate of the pure drug. In vitro dissolution studies have reported the effect of sugar concentration (drug-to-sugar ratios of 4:1, 2:1, 1:1, and 1:4) on drug release. The increase in the sugar concentration increased the drug release. However, none of the sugar-based SDs have reported 100% drug release after 60 min and, therefore, an increase in the sugar content had an insignificant effect [40].
Similar results were reported by Madgulkar et al. [7] when SDs of clotrimazole were prepared using sugars (D-fructose, D-dextrose, and D-maltose). The results indicated a decrease or insignificant change in the saturation solubility and dissolution rate for dextrose, lactose, and maltose, while the opposite was the case for fructose. This phenomenon in sugars was explained by Etman and Naggar [41] as competition for hydrogen bonding with water molecules, which facilitates the solvation of the drug. An increase in sugar concentrations results in having many hydrogen-bonding sites competing against the polar groups of the drug molecules. Another proposed theory indicates that an increase in sugar concentration results in supersaturation, leading to the crystallisation of the sugar and growth in the crystal size [7].
One of the important characteristics of carriers used in the preparation of SDs is their stability in heat [5]. In the studies described above, it was found that the application of sugars in SDs shows limitations based on the occurrence of the Maillard reaction when they are heated to above their melting point, causing the degradation of the sugar. Often, the solid dispersions cannot be heated to melt both the drug and the carrier, resulting in the partial distribution of the drug at the molecular level; hence, a bi-phasic drug release is observed in most cases, and the drug cannot be released rapidly.
In addressing the problem of miscibility at the molecular level, ultrasonication was applied after melting an indomethacin–glucose mix and prior to cooling it to room temperature. This assisted in dispersing the drug and sugar homogeneously, resulting in the formation of two-phase amorphous–amorphous solid dispersions (Class A-A), signified by an amorphous drug dispersed in an amorphous carrier. While dissolution studies showed an eight-fold increase in drug release, pharmacokinetic studies only showed an increase in bioavailability of 1.9-fold. It is worth noting that the resultant SDs remained amorphous for 2 years at room temperature [42]. Of note, ultrasonication might not be the sole reason for the formation of ASDs, as the glass-forming ability of both the drug and sugar plays a significant role [22].
While the fusion and quench-cooling methods have been extensively utilized, they are mostly conducive to forming solid suspensions rather than solid solutions. This inherent limitation constrains the potential of sugars as carriers to significantly enhance drug solubility and dissolution rates. An analysis of the studies reviewed indicates that factors such as a low degradation threshold, a lack of miscibility between the drug and sugar at elevated temperatures, hygroscopicity, and solution-mediated phase transitions represent the principal shortcomings of sugars in SD preparation via the fusion or quench-cooling methods. The recrystallisation of the drug during dissolution can predominantly be attributed to the rapid dissolution of sugar in the solvent due to its high solubility. This leads to the creation of a supersaturated environment and competes for hydrogen-bonding sites with the solvent.
In the preparation of SDs, solvent evaporation is also considered one of the most commonly used methods, particularly when dealing with drug–carrier mixtures where one of the compounds either possesses a high melting point or undergoes degradation when heated [43]. Etoricoxib SDs were prepared with sugar carriers such as lactose and sucrose in 1:1 and 1:5 ratios using the solvent evaporation technique. The drug–sugar mixture is dissolved in ethanol and subsequently heated in a water bath at 60 °C to facilitate solvent removal. An FTIR analysis of these SDs indicated potential intermolecular hydrogen bonding between the S=O group of etoricoxib and the O–H group of the sugar carriers. X-ray diffraction (XRD) and differential scanning calorimetry (DSC) unveiled the absence of characteristic peaks associated with the drug, coupled with the reduced enthalpy of etoricoxib, indicating a shift towards an amorphous state for etoricoxib within the SDs. Notably, unlike the melting or fusion method, the SD systems formed here were Class A-C, denoting the presence of an amorphous drug dispersed in a crystalline carrier matrix. Equilibrium solubility studies indicated an increase in solubility by 1.8- and 1.5-fold for SDs consisting of lactose and sucrose, respectively, in a 1:5 ratio [44].
The hydrochloride salt of amino sugar, glucosamine hydrochloride (G-HCL), was studied for its potential as a hydrophilic carrier in the preparation of SDs for poorly soluble drugs. SDs of CBZ [45] and acyclovir (ACV) [46] were prepared using the solvent evaporation method. In the case of CBZ, SDs were prepared using single (ethanol or acetone) and binary solvent systems mixed with water. The study evaluated the effect of the carrier concentration and the solvent system on CBZ solubility and the dissolution rate. Interestingly, the results indicated that an increase in the carrier concentration did not consistently result in increased dissolution rates when a single solvent system was used. This phenomenon was mainly attributed to the limited solubility of G-HCL within the organic solvents. To corroborate this finding, dissolution profiles of SDs prepared with a binary solvent system (ethanol–water or acetone–water) were investigated. Notably, SDs from the binary solvent systems showed increased dissolution rates with increasing carrier concentration, underscoring the important role the solvent system plays in facilitating the dissolution of both the drug and the carrier. The same trend was also observed for the solubility study results.
SDs of ACV were prepared using ethanol, wherein the drug and carrier were mixed until the solvent completely evaporated. FTIR results indicated that ACV SDs showed potential intermolecular interactions between the N-H group of ACV and the O-H group of G-HCL. Despite the presence of an amine group in CBZ, no intermolecular interactions were reported in the previous study. The difference may be attributed to the use of grinding in the presence of a solvent, which causes the loosening of the molecules at the reaction sites, leading to a chemical change or phase transition [47]. Furthermore, the grinding process also facilitated the reduced particle size of the ACV, resulting in partial amorphization and enhanced aqueous solubility. Comparatively, ACV physical mixtures and SDs showed a significant increase in solubility of 6- and 12-fold, respectively, in contrast with the pure drug. The study also indicated that the increase in the concentration of the carrier only resulted in a mild-to-moderate increase in aqueous solubility, aligning with findings from the various studies mentioned earlier.
In comparison with the fusion method, SDs prepared via solvent evaporation often show evidence of intermolecular interactions, which could explain the absence of the “parachute” effect during dissolution studies. Intermolecular interactions can suppress molecular mobility, thereby inhibiting the formation of the crystal nucleus and recrystallisation [48]. Nevertheless, several challenges were reported, including the limited solubility of sugars in organic solvents and the necessity of removing organic solvents to avoid solvation during storage. Strategies such as employing binary solvent systems yielded positive results to a certain extent, based on the concentration of the sugar. Further studies could delve into the use of binary solvent systems with less polarity difference and the addition of surfactants to enhance the solubility of sugars in organic solvents.
Freeze-drying stands as one of the most widely used techniques for the preparation of amorphous solid dispersions (ASDs) [49]. In addressing the limitations associated with the fusion and solvent evaporation methods, several authors have explored an alternative approach involving SDs with sugar carriers. In two different studies, van Drooge et al. [50,51] investigated the incorporation of lipophilic drugs into sugar glasses via the lyophilization process, employing a solvent mixture consisting of water and tertiary butyl alcohol. Sugar-glass-based ASDs made of diazepam [50], nifedipine, Δ9-tetrahydrocannabinol (THC), and cyclosporine A [51] were successfully prepared using trehalose, sucrose, and two inulins (inulinDP11 and inulinDP23). Firstly, the results of the study underscore the effectiveness of the lyophilization process in transforming lipophilic drugs into amorphous forms, incorporating such drugs into sugar glasses. The dissolution behaviour studied for tablets containing diazepam SDs exhibited anomalous dissolution, characterized by nonlinear and unpredictable drug release patterns in the case of trehalose, sucrose, and inulinDP11. This phenomenon primarily stems from the high aqueous solubility of sugars, leading to the rapid release of the drug and the formation of a supersaturated system. This resulted in diazepam undergoing solution-mediated phase transformation and recrystallisation [50].
This phenomenon was scrutinized by Srinarong et al. [52], who found similar results, wherein the drug loading and solubility of the carrier were identified as pivotal factors in recrystallisation prevention. Notably, the high Tg exhibited by inulins conferred enhanced physical stability compared with trehalose and sucrose. It is worth noting that the conversion of crystalline drugs into their amorphous counterparts is dependent on the drug. This parameter varies from one drug to another, based on the closeness of the Tg to the melting point [51].
Palatinose, α-maltose, and trehalose were employed in the development of completely amorphous sugar-based solid dispersions (SAS-SDs). These systems were prepared by initially subjecting the sugars to freeze-drying, followed by their solubilization in an organic solvent containing the hydrophobic drug. The initial freeze-drying step led to the formation of amorphous sugars, thereby enhancing their solubility in organic solvents. This technique exhibits great promise, as it allows for the solubilization of substantial quantities of sugars, up to 100 mg/mL, in methanol [53]. Subsequently, the resultant mixture is further processed via vacuum-drying [54,55,56] to remove the organic solvent. The resultant material showed no discernible endothermic events during DSC analysis, indicating the conversion of both components into an amorphous form, thereby yielding ASDs. This noteworthy phenomenon was observed for SDs containing drug loadings ranging between 1 to 10% w/w, where a small endothermic event signified the incomplete amorphization of the drug, thus emphasizing the influence of drug loading [55].
SDs investigated in all of the studies [54,55,56] showed a Tg below 40 °C, indicating the potential vulnerability of these systems to instability during storage and solution-mediated phase transitions during the drug dissolution process. This phenomenon was unequivocally substantiated with dissolution studies, where a distinctive “spring and parachute” effect was consistently observed across these investigations. Furthermore, it was noted that the Tg of these systems decreased with an increase in the drug content, attributable to the plasticizing effect of the hydrophobic drug [56]. The decrease in the Tg of the SDs directly relates to the Tg of the pure sugars [55,56]. However, when the SAS-SDs are subjected to thermal treatments above the melting point of the drug followed by rapid cooling (quenching), a distinctive plateau emerges in the subsequent drug dissolution profile [56].
To address the issue of low Tg, Takeda et al. [57] devised a method involving the heat treatment of ASDs formed through the freeze-drying process. Specifically, SDs comprising α-maltose and hydrophobic drugs were prepared via freeze-drying and subjected to heat treatments between 30–120 °C for durations extending up to 120 min. The results of the study indicated that the Tg of the SDs increased with an increase in temperature and the time of heating, thereby showing the direct relationship of these preparation parameters with the SD’s Tg. Moreover, the dissolution studies showed a marked increase in drug release from the heat-treated ASDs in comparison with the untreated ASDs. However, both the SDs showed the typical “spring and parachute” dissolution profiles. It is worth noting that the authors did not report on the stability of the heat-treated SDs, which represents an intriguing avenue for future exploration, especially concerning the change in Tg during storage.
Various alternative methods, such as kneading, centrifugal spinning, and milling, have been reported for the preparation of SDs using sugars. In the case of allopurinol SDs, the kneading method was employed using maltose, sucrose [58], lactose, and mannitol [59] as sugar carriers. In all cases, water was used as a solvent to wet the drug-carrier mixtures (1:1, 1:3, and 1:5), followed by kneading for 45 min, and they were dried and sieved to achieve a powder with an average particle size of 420 µm. The authors noted a significant improvement in drug release during the dissolution studies. However, the effect was primarily observed for the first two intervals (5 and 10 min), while from 15–60 min, the drug release between the SDs and the pure drug did not differ much for any of the ratios. Fourier-transform infrared (FTIR) spectroscopy results showed no interaction between the drug and the sugar, which could be attributed to the low aqueous solubility of allopurinol. In a separate study, Dai et al. [60] prepared co-crystals of allopurinol using liquid-assisted grinding (LAG), where methanol and isopropyl alcohol were used as solvents. As the solubility of drugs and co-formers in common solvents play a pivotal role in intermolecular interactions [8], the use of a single or binary solvent system that can provide moderate-to-high solubility for both the drug and sugar would be interesting to explore.
Saito et al. [61] used the roller compaction method to prepare griseofulvin (GF) SDs with lactose and maltose as carriers. A mixture of GF and sugar carrier at a weight ratio of 1:4 resulted in a markedly reduced particle size after passing through the roller mill for 20 min. The formation of an amorphous mixture was confirmed by the absence of characteristic diffraction peaks during X-ray diffraction analysis. Dissolution studies indicated a 35- to 40-fold increase in drug release for both sugar-based ASDs. This study, however, reported processing problems stemming from the heavy sticking of the sugars to the rollers, an aspect that may be reduced with the use of the ball milling technique. Additionally, the SDs converted into a crystalline state within 24 h when exposed to accelerated storage conditions, indicating instability.
Sucrose-loaded micro-fibrous SDs of olanzapine and piroxicam were prepared using the centrifugal spinning method. The characterization of the microfibers via thermal analysis and hot-stage microscopy confirmed the formation of ASDs by both drugs. Subsequent spectroscopic analysis indicated possible intermolecular interactions between the hydroxyl groups of sucrose and the proton-accepting groups of olanzapine. Dissolution studies, under non-sink conditions, demonstrated rapid drug release, resulting in a three-fold increase in olanzapine and a 1.7-fold increase in piroxicam. The Tg values of these systems were reported to be much higher (about 70 °C), indicating the possible reason for a lack of recrystallisation during dissolution. The study reported achieving high production yield (85%) and drug-loading efficacy (90%) using this method [62].
Despite the several drawbacks reported, recent studies [57,62] have highlighted the potential of using sugars as carriers for SDs to improve the dissolution and solubility of poorly water-soluble drugs. However, further research may be needed to optimize the formulation conditions and assess the long-term stability and practical applicability of sugar-based SD formulations for drug delivery.
2.2. Solid Dispersions of Sugar Alcohol (Polyol) Carriers
Sugar alcohols, also known as polyols, encompass both monosaccharides (erythritol, xylitol, sorbitol, mannitol) and disaccharides (lactitol, isomalt, maltitol). They are formed via the catalytic hydrogenation of carbohydrates, during which, the aldehyde or ketone group is replaced by a hydroxyl group. Currently, there are eight (8) polyols approved by the US FDA, which include erythritol, hydrogenated starch hydrolysates, isomalt, lactitol, maltitol, mannitol, sorbitol, and xylitol [64,65]. Unlike the sugars discussed above, polyols lack a carbonyl group capable of reacting with amino acids, thereby resulting in the Maillard reaction. Polyols are reported to be extremely stable during heat and enzymatic and chemical degradation [66]. In recent times, there has been a burgeoning interest in incorporating polyols into various types of dosage forms such as liquid, oral preparations, lozenges, and tablets. Polyols are highly potent; are low in caloric content; have high nutritional value; and most importantly, are non-carcinogenic [10]. Several authors have reported the use of polyols such as mannitol, sorbitol, and xylitol in the preparation of SDs and their effect on the enhancement of the dissolution rate of poorly soluble drugs. In order to realize the potential of polyols to be used as carriers in the successful preparation of SDs, it is important to review the physicochemical properties associated with these compounds. A summary of the various polyols and their physicochemical properties is presented in .
3. Co-Amorphous and Co-Crystalline Systems
Co-amorphous and co-crystalline systems are an emerging formulation approach involving combining the drug with hydrophilic, small-molecular-weight compounds such as amino acids, carboxylic acids, and sugars in the preparation of drug–excipient systems in order to improve drug solubility and bioavailability. Studies have indicated that these drug–excipient systems yield improved dissolution rates and stability compared with their respective crystalline or amorphous counterparts. These improvements are facilitated through intermolecular interactions such as hydrogen bonding and π–π interactions [120,121]. Co-amorphous systems stabilize the amorphous forms through strong and specific molecular interactions between the drug and partner molecule and by increasing the glass transition temperature. In this system, amorphous forms are quite stable, as they need a stronger effort to go back to their crystalline state, in that they first have to break the molecular interactions and then rearrange their molecules to form crystalline nuclei [122]. Also, the availability of a wide range of suitable co-formers, compared with counterions for salt formation, makes this approach attractive [123,124]. By selecting an appropriate co-former, co-crystals offer tailormade solutions to various solid-state problems, such as physicochemical stability, including photostability, hygroscopicity, permeability, compressibility, flow properties, solubility, bioavailability, pharmacokinetic properties, etc., without altering the molecular structure and pharmacological properties [123,125].
It is worthwhile to note that no studies have explored the formation of co-amorphous systems using sugars or polyols; only one study has surfaced pertaining to co-crystals with polyols. Arafa et al. [16] reported xylitol as a potential co-former in the preparation of felodipine co-crystals. The authors reported successful co-crystal formation with a 1:2 ratio of felodipine to xylitol using a wet co-grinding method. The characterization of the co-crystals with DSC showed two characteristic melting peaks in the thermogram, corresponding to both the carrier and the drug. Typically, co-crystals are single-phase systems [126] that are characterised by the presence of a single endothermic peak between the melting temperatures of the individual compounds [8,127]. The lack of single-crystal XRD data also makes it difficult to confirm the formation of co-crystals with xylitol. With the supramolecular synthon approach, polyols have the potential to form co-crystals. The propensity of sugars to engage in a multitude of hydrogen bonds has established their significance in supramolecular chemistry. A heterosynthon between the hydroxyl groups of polyols and drugs consisting of functional groups such as ether could conceivably result in the formation of a co-crystal. However, this prospect warrants further research to definitively establish it.
4. Natural Deep Eutectic Solvents
In the quest to address the inherent toxicity of organic solvents used in the food, chemical, and pharmaceutical industries, alternative green and biodegradable solvents such as ionic liquids (ILs), deep eutectic solvents (DESs), and natural deep eutectic solvents (NADESs) have emerged. While the “greenness” of ILs is often questioned, DESs have been extensively studied for their pharmaceutical applications to improve the solubility and bioavailability of poorly soluble drugs. However, their safety is also questionable, as not much data from in vivo and long-term stability are available [128,129]. NADESs are a mixture of two or more components from natural sources, including sugars, sugar alcohols, poly alcohols, amino acids, and organic acids [129,130]. NADESs are considered superior to DESs because of their unique physicochemical properties, such as low volatility, an ability to remain in the liquid state in sub-zero temperatures, biodegradability, and ease of preparation [131]. Despite the advantages reported, its applications in pharmaceutical research have not gained full momentum yet.
In two different studies by Jeliński et al. [131,132], various sugars (fructose, glucose, maltose, sucrose) and polyols (xylitol, sorbitol) were investigated for the solubility enhancement of sulphanilamide and curcumin, respectively. NADESs were prepared with choline chloride as a primary constituent, and one of the sugars or polyols was mixed with it in different molar ratios. All the resultant NADESs showed a two-fold increase in solubility for sulphanilamide and a significant increase in curcumin, with the lowest being 1000-fold. Maugeri and De Marı’a [14] reported the neutral pH for sugar-based deep eutectic solvents, which is of considerable importance in pharmaceutical applications, mainly in terms of stability. In addition to these studies, Dai et al. [133] reported the possibility of preparing NADESs with different combinations of sugars and polyols. Though there is limited literature available on the pharmaceutical applications of NADESs, their applications as extraction media are well documented [129,134]. The fundamental knowledge obtained from these studies can be extrapolated to address the solubility challenges of various BCS Class II and IV drugs.
5. Conclusions and Future Perspectives
There is growing interest in utilizing low-molecular-weight, water-soluble carriers for enhancing the solubility and dissolution rates of poorly soluble drugs. Initially, sugars were the preferred carriers in the formulation of first-generation SDs given their low molecular weight and hydrophilic nature. However, their application is met with several challenges, primarily their low thermal stability, limited miscibility, and hygroscopic nature. As the majority of the SDs formed using sugars result in crystalline suspensions, the drug release from these formulations has been found to be much slower compared with amorphous forms. The use of polyols has effectively addressed these challenges and has proved to be a better choice of carriers over sugars. The effectiveness of these carriers in enhancing the solubility of drugs is dependent on a number of factors, including physicochemical properties such as the melting point, Tg, the miscibility of drugs in the carrier, etc. The preparation method also plays a significant role, with the fusion technique showing more success over solvent evaporation, mainly because of the limited solubility of the sugars and polyols in organic solvents.
The use of ultrasonication and freeze-drying have shown potential in improving miscibility and the formation of ASDs with both sugars and polyols; however, these ASDs have been found to be unstable. Strategies such as the preparation of ternary solid dispersions by including amino acids, synthetic hydrophilic polymers, or poloxamers have proven to be highly successful in obtaining stable ASDs. Second-generation SDs, which can achieve this without the inclusion of sugars or polyols, come with unique challenges, such as requiring high concentrations of polymers, which can lead to viscosity increases during dissolution and imparting their characteristic taste in the case of amino acids. Considering that sugars and polyols are primarily used in the pharmaceutical industry for their taste-masking properties and solubilizing capacity, they can be the preferred choice of carriers in the preparation of ternary solid dispersions, which is the current direction of research. Novel techniques such as NanoCrySP™ used to prepare nanocrystalline solid dispersions with polyols should also be explored, as they also show very promising results.
In this review, it is evident that polyols such as xylitol, erythritol, isomalt, and maltitol are relatively underexplored. The preliminary observations suggest that they have potential applications in formulating stable SDs, and the good glass-forming abilities of isomalt and maltitol are of particular interest for preparing ASDs. In addition to this, preparing drug–excipient-based co-amorphous and co-crystalline systems has seen limited exploration, with only a few low-molecular-weight excipients being explored. Given the potential of these systems to improve the dissolution characteristics of poorly soluble drugs, the proven ability of some low-molecular-weight artificial sugars, such as saccharin and sucralose, provides the background for exploring low-molecular-weight polyols as potential excipients. Moreover, the use of natural sugars and polyols in the formulation of NADES-based drug delivery systems presents great research opportunities.
References
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems Shweta. ISRN Pharm. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Ainurofiq, A.; Putro, D.S.; Ramadhani, D.A.; Putra, G.M.; Do Espirito Santo, L.D.C. A Review on Solubility Enhancement Methods for Poorly Water-Soluble Drugs. J. Rep. Pharm. Sci. 2021, 10, 137–147. [Google Scholar] [CrossRef]
- Pešić, N.; Dapčević, A.; Ivković, B.; Kachrimanis, K.; Mitrić, M.; Ibrić, S.; Medarević, D. Potential Application of Low Molecular Weight Excipients for Amorphization and Dissolution Enhancement of Carvedilol. Int. J. Pharm. 2021, 608, 121033. [Google Scholar] [CrossRef] [PubMed]
- Tekade, A.R.; Yadav, J.N. A Review on Solid Dispersion and Carriers Used Therein for Solubility Enhancement of Poorly Water Soluble Drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef]
- Kumar Bandaru, R.; Rout, S.R.; Kenguva, G.; Gorain, B.; Alhakamy, N.A.; Kesharwani, P.; Dandela, R. Recent Advances in Pharmaceutical Cocrystals: From Bench to Market. Front. Pharmacol. 2021, 12, 780582. [Google Scholar] [CrossRef]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as Solid Dispersion Carrier to Improve Solubility and Dissolution of the BCS Class II Drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef]
- Thayyil, A.R.; Juturu, T.; Nayak, S.; Kamath, S. Pharmaceutical Co-Crystallization: Regulatory Aspects, Design, Characterization, and Applications. Adv. Pharm. Bull. 2020, 10, 203–212. [Google Scholar] [CrossRef]
- White, J.R. Sugar. Clin. Diabetes 2018, 36, 74–76. [Google Scholar] [CrossRef]
- Priya, K.; Gupta, V.R.M.; Srikanth, K. Natural Sweeteners: A Complete Review. J. Pharm. Res. 2011, 4, 2034–2039. [Google Scholar]
- Bazeed, A.Y.; Nouh, A.; Essa, E.A.; El Maghraby, G.M. Hydrophilic Sugars for Enhancing Dissolution Rate of Cilostazol: Effect of Wet Co-Processing. Pharm. Sci. 2021, 27, 111–120. [Google Scholar] [CrossRef]
- Bindhani, S.; Mohapatra, S. Recent Approaches of Solid Dispersion: A New Concept toward Oral Bioavailability. Asian J. Pharm. Clin. Res. 2018, 11, 72–78. [Google Scholar] [CrossRef]
- Silva, L.P.; Fernandez, L.; Conceiçao, J.H.F.; Martins, M.A.R.; Sosa, A.; Ortega, J.; Pinho, S.P.; Coutinho, J.A.P. Design and Characterization of Sugar-Based Deep Eutectic Solvents Using Conductor-like Screening Model for Real Solvents. ACS Sustain. Chem. Eng. 2018, 6, 10724–10734. [Google Scholar] [CrossRef]
- Maugeri, Z.; De María, P.D. Novel Choline-Chloride-Based Deep-Eutectic-Solvents with Renewable Hydrogen Bond Donors: Levulinic Acid and Sugar-Based Polyols. RSC Adv. 2012, 2, 421–425. [Google Scholar] [CrossRef]
- Mousa, T.M.; Donia, A.A.; El Maghraby, G.M. Co-Crystallization of Sofosbuvir with Sugars for Enhanced Dissolution Rate. Indones. J. Pharm. 2023, 34, 140–152. [Google Scholar] [CrossRef]
- Arafa, M.F.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Xylitol as a Potential Co-Crystal Co-Former for Enhancing Dissolution Rate of Felodipine: Preparation and Evaluation of Sublingual Tablets. Pharm. Dev. Technol. 2018, 23, 454–463. [Google Scholar] [CrossRef]
- Sun, T.; Watson, S. Sorbitol/Dexlansoprazole Co-Crystals and Method for Making Same. U.S. Patent 8,318,943 B1, 27 November 2012. Available online: https://patentimages.storage.googleapis.com/8b/19/bb/4167c62761fffc/US8318943.pdf (accessed on 26 July 2023).
- Simperler, A.; Kornherr, A.; Chopra, R.; Bonnet, P.A.; Jones, W.; Motherwell, W.D.S.; Zifferer, G. Glass Transition Temperature of Glucose, Sucrose, and Trehalose: An Experimental and in Silico Study. J. Phys. Chem. B 2006, 110, 19678–19684. [Google Scholar] [CrossRef]
- Malkawi, R.; Malkawi, W.I.; Al-Mahmoud, Y.; Tawalbeh, J. Current Trends on Solid Dispersions: Past, Present, and Future. Adv. Pharmacol. Pharm. Sci. 2022, 2022, 5916013. [Google Scholar] [CrossRef]
- Tambe, S.; Jain, D.; Meruva, S.K.; Rongala, G.; Juluri, A.; Nihalani, G.; Mamidi, H.K.; Nukala, P.K.; Bolla, P.K. Recent Advances in Amorphous Solid Dispersions: Preformulation, Formulation Strategies, Technological Advancements and Characterization. Pharmaceutics 2022, 14, 2203. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the Development of Amorphous Solid Dispersions: The Role of Polymeric Carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous Solid Dispersions: An Update for Preparation, Characterization, Mechanism on Bioavailability, Stability, Regulatory Considerations and Marketed Products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, Y.L.; Parashar, B.; Ostwal, P.P.; Jain, M.S. Solid Dispersion: Solubility Enhancement for Poorly Water Soluble Drug. Res. J. Pharm. Technol. 2012, 5, 190–197. [Google Scholar]
- Meng, F.; Gala, U.; Chauhan, H. Classification of Solid Dispersions: Correlation to (i) Stability and Solubility (Ii) Preparation and Characterization Techniques. Drug Dev. Ind. Pharm. 2015, 41, 1401–1415. [Google Scholar] [CrossRef]
- Attia, M.S.; Hasan, A.A.; Ghazy, F.E.S.; Gomaa, E. Solid Dispersion as a Technical Solution to Boost the Dissolution Rate and Bioavailability of Poorly Water-Soluble Drugs. Indian J. Pharm. Educ. Res. 2021, 55, s327–s339. [Google Scholar] [CrossRef]
- Nikam, V.K.; Shete, S.K.; Khapare, J.P. Most Promising Solid Dispersion Technique of Oral Dispersible Tablet. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 1–16. [Google Scholar] [CrossRef]
- Sharma, K.S.; Sahoo, J.; Agrawal, S.; Kumari, A. Solid Dispersions: A Technology for Improving Bioavailability. J. Anal. Pharm. Res. Rev. 2019, 8, 127–133. [Google Scholar] [CrossRef]
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical Cocrystals: Regulatory and Strategic Aspects, Design and Development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Devhare, L.D.; Kore, P.K. A Recent Review on Bioavailability and Solubility Enhancement of Poorly Soluble Drugs by Physical and Chemical Modifications. Res. Chron. Health Sci. 2016, 2, 299–308. [Google Scholar]
- Lewis, S.; Udupa, N. Solid Dispersions: A Review. Pak. J. Pharm. Sci. 2009, 22, 234–246. [Google Scholar]
- Singh, A.; Van den Mooter, G. Spray Drying Formulation of Amorphous Solid Dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Soyata, A.; Kenti, K.; Sutoro, M.; Sagita, N. Impact of Preparation Method in Co-Amorphous System. Sci. Pharm. 2022, 1, 47–55. [Google Scholar] [CrossRef]
- Vo, C.-L.N.; Park, C.; Lee, B.J. Current Trends and Future Perspectives of Solid Dispersions Containing Poorly Water-Soluble Drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V.; Yanchick, V.A.; Maness, D.D. Dissolution Rates of Corticosteroids Utilizing Sugar Glass Dispersions. J. Pharm. Sci. 1977, 66, 494–497. [Google Scholar] [CrossRef]
- Allen, L.V.; Levinson, R.S.; De Martono, D. Dissolution Rates of Hydrocortisone and Prednisone Utilizing Sugar Solid Dispersion Systems in Tablet Form. J. Pharm. Sci. 1978, 67, 979–981. [Google Scholar] [CrossRef]
- Ghanem, A.; Meshali, M.; Ibraheem, Y. Dissolution Rates of Sulfamethoxazole Utilizing Sugar Glass Dispersions. J. Pharm. Pharmacol. 1980, 32, 675–677. [Google Scholar] [CrossRef]
- Bouchard, A.; Hofland, G.W.; Witkamp, G.J. Properties of Sugar, Polyol, and Polysaccharide Water-Ethanol Solutions. J. Chem. Eng. Data 2007, 52, 1838–1842. [Google Scholar] [CrossRef]
- Hirasawa, N.; Okamoto, H.; Danjo, Z. Lactose as a Low Molecular Weight Carrier of Solid Dispersions for Carbamazepine and Ethenzamide. Chem. Pharm. Bull. 1999, 3, 417–420. [Google Scholar] [CrossRef]
- Saffoon, N.; Jhanker, Y.M.; Huda, N.H. Dissolution Profile of Ibuprofen Solid Dispersion Prepared with Cellulosic Polymers and Sugar by Fusion Method. Stamford J. Pharm. Sci. 2011, 4, 31–37. [Google Scholar] [CrossRef]
- Etman, M.A.; Naggar, V.F. Thermodynamics of Paracetamol Solubility in Sugar-Water Cosolvent Systems. Int. J. Pharm. 1990, 58, 177–184. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; Silva, T.L.; Castro, S.; Lara-díaz, V.J.; Castorena-torres, F. Two-Phase Amorphous-Amorphous Solid Drug Dispersion with Enhanced Stability, Solubility and Bioavailability Resulting from Ultrasonic Dispersion of an Immiscible System. Eur. J. Pharm. Biopharm. 2017, 119, 243–252. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Das, A.; Nayak, A.K.; Mohanty, B.; Panda, S. Solubility and Dissolution Enhancement of Etoricoxib by Solid Dispersion Technique Using Sugar Carriers. ISRN Pharm. 2011, 2011, 819765. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamidi, H.; Edwards, A.A.; Mohammad, M.A.; Nokhodchi, A. To Enhance Dissolution Rate of Poorly Water-Soluble Drugs: Glucosamine Hydrochloride as a Potential Carrier in Solid Dispersion Formulations. Colloids Surf. B Biointerfaces 2010, 76, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Telange, D.R.; Bhagat, S.B.; Patil, A.T.; Umekar, M.J.; Pethe, A.M.; Raut, N.A.; Dave, V.S. Glucosamine HCL-Based Solid Dispersions to Enhance the Biopharmaceutical Properties of Acyclovir. J. Excipients Food Chem. 2019, 10, 65–81. [Google Scholar]
- Pagola, S. Outstanding Advantages, Current Drawbacks, and Significant Recent Developments in Mechanochemistry: A Perspective View. Crystals 2023, 13, 124. [Google Scholar] [CrossRef]
- Budiman, A.; Nurfadilah, N.; Muchtaridi, M.; Sriwidodo, S.; Aulifa, D.L.; Rusdin, A. The Impact of Water-Soluble Chitosan on the Inhibition of Crystal Nucleation of Alpha-Mangostin from Supersaturated Solutions. Polymers 2022, 14, 4370. [Google Scholar] [CrossRef]
- Siow, C.R.S.; Wan Sia Heng, P.; Chan, L.W. Application of Freeze-Drying in the Development of Oral Drug Delivery Systems. Expert. Opin. Drug Deliv. 2016, 13, 1595–1608. [Google Scholar] [CrossRef]
- Van Drooge, D.J.; Hinrichs, W.L.J.; Frijlink, H.W. Anomalous Dissolution Behaviour of Tablets Prepared from Sugar Glass-Based Solid Dispersions. J. Control. Release 2004, 97, 441–452. [Google Scholar] [CrossRef]
- Van Drooge, D.J.; Hinrichs, W.L.J.; Frijlink, H.W. Incorporation of Lipophilic Drugs in Sugar Glasses by Lyophilization Using a Mixture of Water and Tertiary Butyl Alcohol as Solvent. J. Pharm. Sci. 2004, 93, 713–725. [Google Scholar] [CrossRef]
- Srinarong, P.; Kouwen, S.; Visser, M.R.; Hinrichs, W.L.J.; Frijlink, H.W. Effect of Drug-Carrier Interaction on the Dissolution Behavior of Solid Dispersion Tablets. Pharm. Dev. Technol. 2010, 15, 460–468. [Google Scholar] [CrossRef]
- Nikghalb, L.A.; Singh, G.; Singh, G.; Kahkeshan, K.F. Solid Dispersion: Methods and Polymers to Increase the Solubility of Poorly Soluble Drugs. J. Appl. Pharm. Sci. 2012, 2, 170–175. [Google Scholar] [CrossRef]
- Satoh, T.; Hidaka, F.; Miyake, K.; Yoshiyama, N.; Takeda, K.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Surfactant-Free Solid Dispersion of Fat-Soluble Flavour in an Amorphous Sugar Matrix. Food Chem. 2016, 197, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Gotoda, Y.; Hirota, D.; Hidaka, F.; Sato, T.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Surfactant-Free Solid Dispersions of Hydrophobic Drugs in an Amorphous Sugar Matrix Dried from an Organic Solvent. Mol. Pharm. 2017, 14, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Sekitoh, T.; Okamoto, T.; Fujioka, A.; Tramis, O.; Takeda, K.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Sole-Amorphous-Sugar-Based Solid Dispersion of Curcumin and the Influence of Formulation Composition and Heat Treatment on the Dissolution of Curcumin. Dry. Technol. 2021, 39, 2065–2074. [Google Scholar] [CrossRef]
- Takeda, K.; Sekitoh, T.; Fujioka, A.; Yamamoto, K.; Okamoto, T.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Physical Stability of an Amorphous Sugar Matrix Dried from Methanol as an Amorphous Solid Dispersion Carrier and the Influence of Heat Treatment. J. Pharm. Sci. 2019, 108, 2056–2062. [Google Scholar] [CrossRef]
- Kaljoriya, H.; Mann, M. Sugar Carriers. Int. J. Pharm. Life Sci. 2021, 12, 5–10. [Google Scholar]
- Jyoti, J.; Shikha, D.A. Solubility Enhancement of Allopurinol by Solid Dispersion Using Sugar Carriers. Int. J. Curr. Res. 2019, 11, 6524–6529. [Google Scholar]
- Dai, X.L.; Yao, J.; Wu, C.; Deng, J.H.; Mo, Y.H.; Lu, T.B.; Chen, J.M. Solubility and Permeability Improvement of Allopurinol by Cocrystallization. Cryst. Growth Des. 2020, 20, 5160–5168. [Google Scholar] [CrossRef]
- Saito, M.; Ugajin, T.; Nozawa, Y.; Sadzuka, Y.; Miyagishima, A.; Sonobe, T. Preparation and Dissolution Characteristics of Griseofulvin Solid Dispersions with Saccharides. Int. J. Pharm. 2002, 249, 71–79. [Google Scholar] [CrossRef]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Craig, D.Q.M. Development of Micro-Fibrous Solid Dispersions of Poorly Water-Soluble Drugs in Sucrose Using Temperature-Controlled Centrifugal Spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.A.; Habib, F.S. Dissolution Rates of Carbamazepine and Nitrazepam Utilizing Sugar Solid Dispersion System. Drug Dev. Ind. Pharm. 1985, 11, 1957–1969. [Google Scholar] [CrossRef]
- Lenhart, A.; Chey, W.D. A Systematic Review of the Effects of Polyols on Gastrointestinal Health and Irritable Bowel Syndrome. Adv. Nutr. 2017, 8, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, M. Sugar Alcohols. In Encyclopedia of Food Chemistry; Melton, L., Shahidi, F., Varelis, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, pp. 265–275. ISBN 9780128140451. [Google Scholar]
- Embuscado, M.E. Polyols. In Optimising Sweet Taste in Foods; Spillane, W.J., Ed.; Woodhead Publishing: Sawston, UK, 2006; pp. 153–174. ISBN 9781845690083. [Google Scholar]
- Hadjikinova, R.; Marudova, M.; Hadjikinova, R.; Marudova, M. Thermal Behaviour of Confectionary Sweeteners’ Blends. Bulg. Chem. Commun. 2016, 48, 446–450. [Google Scholar]
- Zumbé, A.; Lee, A.; Storey, D. Polyols in Confectionery: The Route to Sugar-Free, Reduced Sugar and Reduced Calorie Confectionery. Br. J. Nutr. 2001, 85, S31–S45. [Google Scholar] [CrossRef]
- Talja, R.A.; Roos, Y.H. Phase and State Transition Effects on Dielectric, Mechanical, and Thermal Properties of Polyols. Thermochim. Acta 2001, 380, 109–121. [Google Scholar] [CrossRef]
- Langer, M.; Höltje, M.; Urbanetz, N.A.; Brandt, B.; Höltje, H.D.; Lippold, B.C. Investigations on the Predictability of the Formation of Glassy Solid Solutions of Drugs in Sugar Alcohols. Int. J. Pharm. 2003, 252, 167–179. [Google Scholar] [CrossRef]
- Chawla, G.; Bansal, A.K. Improved Dissolution of a Poorly Water Soluble Drug in Solid Dispersions with Polymeric and Non-Polymeric Hydrophilic Additives. Acta Pharm. 2008, 58, 257–274. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Hu, A.; Nie, T.; Cheng, Z.; Liu, W. A Critical Review on Engineering of D-Mannitol Crystals: Properties, Applications, and Polymorphic Control. Crystals 2022, 12, 1080. [Google Scholar] [CrossRef]
- Punitha, S.; Bn, V.H.; Karthikeyan, D. Enhancement of Celecoxib Solubility by Solid Disperson Using Mannitol. Int. J. Pharm. Pharm. Sci. 2010, 2, 4–6. [Google Scholar]
- Bahmani, K.; Singla, Y. Enhanced Solubility of Antihypertensive Drug Using Hydrophilic Carrier-Based Potent Solid Dispersion Systems. Int. J. Pharm. Res. Technol. 2019, 9, 24–37. [Google Scholar]
- Yadav, P.S.; Kumar, V.; Pratap, U.; Raj, H.; Mazumder, B. Physicochemical Characterization and in Vitro Dissolution Studies of Solid Dispersions of Ketoprofen with PVP K30 and D-Mannitol. Saudi Pharm. J. 2013, 21, 77–84. [Google Scholar] [CrossRef]
- Singh, G.; Chhabra, G.; Pathak, K. Dissolution Behavior and Thermodynamic Stability of Fused-Sugar Dispersions of a Poorly Water-Soluble Drug. Dissolution Technol. 2011, 18, 62–70. [Google Scholar] [CrossRef]
- Shittu, A.O.; Njinga, N.S.; Orshio, S.D. Development and Characterization of Ibuprofen Solid Dispersion for Solubility and Dissolution Improvement Using a Binary Carrier System Consisting of D-Mannitol—Polyethylene Glycol 6000. Niger. J. Pharm. 2022, 56, 109–118. [Google Scholar]
- Zajc, N.; Obreza, A.; Bele, M.; Srčič, S. Physical Properties and Dissolution Behaviour of Nifedipine/Mannitol Solid Dispersions Prepared by Hot Melt Method. Int. J. Pharm. 2005, 291, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Patel, R.; Shah, H.; Purohit, S.; Pawar, M.; Pathan, A. Solubility Enhancement of Azithromycin by Solid Dispersion Technique Using Mannitol and β-Cyclodextrin. Acta Sci. Pharm. Sci. 2022, 5, 48–54. [Google Scholar] [CrossRef]
- Walke, P.S.; Khairnar, P.S.; Narkhede, M.R.; Nehete, J.Y. Solubility Enhancement of Diacerein by Mannitol Solid Dispersons. Int. J. Pharm. Pharm. Sci. 2011, 3, 261–264. [Google Scholar]
- Krishnamoorthy, V.; Priya, V.; Prasad, R. Physicochemical Characterization and in Vitro Dissolution Behavior of Olanzapine-Mannitol Solid Dispersions. Braz. J. Pharm. Sci. 2012, 48, 243–255. [Google Scholar] [CrossRef]
- Okonogi, S.; Oguchi, T.; Yonemochi, E.; Puttipipatkhachorn, S.; Yamamoto, K. Improved Dissolution of Ofloxacin via Solid Dispersion. Int. J. Pharm. 1997, 156, 175–180. [Google Scholar] [CrossRef]
- Dubey, A.; Road, R.; Pradesh, M. Enhancement of Aqueous Solubility and Dissolution of Telmisartan. Int. J. Pharm. Sci. Res. 2014, 5, 4478–4485. [Google Scholar] [CrossRef]
- Neupane, S.; Thapa, C. Formulation and Enhancement of Dissolution Rate of Poorly Aqueous Soluble Drug Aceclofenac by Solid Dispersion Method: In Vitro Study. Afr. J. Pharm. Pharmacol. 2020, 14, 1–8. [Google Scholar] [CrossRef]
- Rao, Y.S.; Vijaya, L.; Varalakshmi, T.; Chandana, R.; Chowdary, K.P.R. Formulation and Evaluation of Carvedilol Solid Dispersions for Dissolution Rate Enhancement. Int. J. Adv. Pharm. Biol. Chem. 2012, 1, 489–495. [Google Scholar]
- Kanaze, F.I.; Kokkalou, E.; Niopas, I.; Georgarakis, M.; Stergiou, A.; Bikiaris, D. Thermal Analysis Study of Flavonoid Solid Dispersions Having Enhanced Solubility. J. Therm. Anal. Calorim. 2006, 83, 283–290. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Verma, P.; Sen, S. Studies on Clozapine-Mannitol Solid Dispersions, Physico Chemical Characterization and Characterization and Evaluation. Turk. J. Pharm. Sci. 2013, 10, 109–124. [Google Scholar]
- Duret, C.; Wauthoz, N.; Sebti, T.; Vanderbist, F.; Amighi, K. Solid Dispersions of Itraconazole for Inhalation with Enhanced Dissolution, Solubility and Dispersion Properties. Int. J. Pharm. 2012, 428, 103–113. [Google Scholar] [CrossRef]
- Kauppinen, A.; Broekhuis, J.; Grasmeijer, N.; Tonnis, W.; Ketolainen, J. Efficient Production of Solid Dispersions by Spray Drying Solutions of High Solid Content Using a 3-Fluid Nozzle. Eur. J. Pharm. Biopharm. 2018, 123, 50–58. [Google Scholar] [CrossRef]
- El-maradny, H.; Saab, M. Spray-Dried Co-Amorphous Tadalafil Ternary Mixtures: A Promising Platform towards the Enhancement of Solubility and Bioavailability. Braz. J. Pharm. Sci. 2022, 58, 1–14. [Google Scholar]
- Thakur, P.S.; Thakore, S.D.; Bansal, A.K. Role of Surface Characteristics of Mannitol in Crystallization of Feno Fi Brate During Spray Drying. J. Pharm. Sci. 2020, 109, 1105–1114. [Google Scholar] [CrossRef]
- Thakral, S.; Sonje, J.; Munjal, B.; Bhatnagar, B.; Suryanarayanan, R. Mannitol as an Excipient for Lyophilized Injectable Formulations. J. Pharm. Sci. 2023, 112, 19–35. [Google Scholar] [CrossRef]
- Verma, U.; Naik, J.B.; Mokale, V.J. Preparation of Freeze-Dried Solid Dispersion Powder Using Mannitol to Enhance Solubility of Lovastatin and Development of Sustained Release Tablet Dosage Form. Am. J. Pharm. Sci. Nanotechnol. 2014, 1, 11–26. [Google Scholar]
- Kulthe, V.; Chaudhari, P.; Aboul-Enein, H. Freeze-Dried Amorphous Dispersions for Solubility Enhancement of Thermosensitive API Having Low Molecular Lipophilicity. Drug Res. 2014, 64, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Muehlenfeld, C.; Kann, B.; Windbergs, M.; Thommes, M. Solid Dispersions Prepared by Continuous Cogrinding in an Air Jet Mill. J. Pharm. Sci. 2013, 102, 4132–4139. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, I.C.S.; Bazzo, G.C.; Zétola, M.; Stulzer, H.K.; Soares, L.; Pezzini, B.R. Ball-Milled Valsartan and Its Combination with Mannitol: The Case of Drug Polyamorphism. J. Therm. Anal. Calorim. 2022, 147, 8765–8777. [Google Scholar] [CrossRef]
- Zaini, E.; Umar, S.; Firdaus, N. Improvement of Dissolution Rate of Valsartan by Solid Dispersion System Using D(−) Mannitol. Asian J. Pharm. Clin. Res. 2017, 10, 288–290. [Google Scholar] [CrossRef]
- Deis, R.C.; Kearsley, M.W. Sorbitol and Mannitol. In Sweeteners and Sugar Alternatives in Food Technology; Kay O’Donnell, M.W.K., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 331–346. ISBN 9780470659687. [Google Scholar]
- Dash, R.P.; Srinivas, N.R.; Babu, R.J. Use of Sorbitol as Pharmaceutical Excipient in the Present Day Formulations–issues and Challenges for Drug Absorption and Bioavailability. Drug Dev. Ind. Pharm. 2019, 45, 1421–1429. [Google Scholar] [CrossRef]
- Hasan, A.; El, A.; Elghany, M.A.; Sabry, S. Design and Characterization of Intra-Oral Fast Dissolving Tablets Containing Diacerein-Solid Dispersion. J. Appl. Pharm. Sci. 2020, 10, 44–53. [Google Scholar] [CrossRef]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid Dispersion as an Approach for Bioavailability Enhancement of Poorly Water-Soluble Drug Ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef]
- Kaialy, W.; Maniruzzaman, M.; Shojaee, S.; Nokhodchi, A. Antisolvent Precipitation of Novel Xylitol-Additive Crystals to Engineer Tablets with Improved Pharmaceutical Performance. Int. J. Pharm. 2014, 477, 282–293. [Google Scholar] [CrossRef]
- Sirenius, I.; Krogerus, V.E.; Leppänen, T. Dissolution Rate of P-aminobenzoates from Solid Xylitol Dispersions. J. Pharm. Sci. 1979, 68, 791–792. [Google Scholar] [CrossRef]
- Mummaneni, V.; Vasavada, R.C. Solubilization and Dissolution of Famotidine from Solid Glass Dispersions of Xylitol. Int. J. Pharm. 1990, 66, 71–77. [Google Scholar] [CrossRef]
- Pawar, J.N.; Fule, R.A.; Maniruzzaman, M.; Amin, P.D. Solid Crystal Suspension of Efavirenz Using Hot Melt Extrusion: Exploring the Role of Crystalline Polyols in Improving Solubility and Dissolution Rate. Mater. Sci. Eng. C 2017, 78, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Semjonov, K.; Kogermann, K.; Laidmäe, I.; Antikainen, O.; Strachan, C.J.; Ehlers, H.; Yliruusi, J.; Heinämäki, J. The Formation and Physical Stability of Two-Phase Solid Dispersion Systems of Indomethacin in Supercooled Molten Mixtures with Different Matrix Formers. Eur. J. Pharm. Sci. 2017, 97, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Narala, S.; Komanduri, N.; Nyavanandi, D.; Adel, A.; Youssef, A.; Mandati, P.; Alzahrani, A.; Kolimi, P.; Narala, N. Hard Gelatin Capsules Containing Hot Melt Extruded Solid Crystal Suspension of Carbamazepine for Improving Dissolution: Preparation and in Vitro Evaluation. J. Drug Deliv. Sci. Technol. 2023, 82, 104384. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.J.; Jeya, M.; Kim, I.W.; Lee, J.K. Biotechnological Production of Erythritol and Its Applications. Appl. Microbiol. Biotechnol. 2010, 86, 1017–1025. [Google Scholar] [CrossRef]
- Mazi, T.A.; Stanhope, K.L. Erythritol: An In-Depth Discussion of Its Potential to Be a Beneficial Dietary Component. Nutrients 2023, 15, 204. [Google Scholar] [CrossRef]
- Tyapkova, O.; Bader-Mittermaier, S.; Schweiggert-Weisz, U. Factors Influencing Crystallization of Erythritol in Aqueous Solutions: A Preliminary Study. J. Food Res. 2012, 1, 207–217. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Hattori, Y.; Otsuka, M. Characterization of Ternary Amorphous Solid Dispersion Containing Hypromellose Phthalate and Erythritol Prepared by Hot Melt Extrusion Using Melting Point Depression. J. Drug Deliv. Sci. Technol. 2020, 58, 101797. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Y.; Yang, W.; Liu, Y.; Liang, M. Solubility of Erythritol in Methanol, Ethanol and Methanol+ethanol: Experimental Measurement and Thermodynamic Modeling. Fluid Phase Equilib. 2013, 360, 134–138. [Google Scholar] [CrossRef]
- Msomi, N.Z.; Erukainure, O.L.; Islam, M.S. Suitability of Sugar Alcohols as Antidiabetic Supplements: A Review. J. Food Drug Anal. 2021, 29, 1–14. [Google Scholar] [CrossRef]
- Ghanavati, R.; Taheri, A.; Homayouni, A. Anomalous Dissolution Behavior of Celecoxib in PVP/Isomalt Solid Dispersions Prepared Using Spray Drier. Mater. Sci. Eng. C 2017, 72, 501–511. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Khalili, N.; Zangiabadi, F.; Homayouni, A. Preparation, Characterization and Stability Studies of Glassy Solid Dispersions of Indomethacin Using PVP and Isomalt as Carriers. Iran. J. Basic Med. Sci. 2012, 15, 820–832. [Google Scholar] [PubMed]
- Moura Ramos, J.J.; Viciosa, M.T.; Diogo, H.P. Thermal Behaviour of Two Anti-Inflammatory Drugs (Celecoxib and Rofecoxib) and Slow Relaxation Dynamics in Their Amorphous Solid State. Comparison between the Dynamic Fragility Obtained by Dielectric Spectroscopy and by Thermostimulated Currents. Mol. Phys. 2019, 117, 644–660. [Google Scholar] [CrossRef]
- Svoboda, R.; Košťálová, D.; Krbal, M.; Komersová, A. Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules 2022, 27, 5668. [Google Scholar] [CrossRef] [PubMed]
- Kearsley, M.W.; Deis, R.C. Maltitol Powder. In Sweeteners and Sugar Alternatives in Food Technology; Wiley: Hoboken, NJ, USA, 2012; pp. 295–308. ISBN 9780470659687. [Google Scholar]
- Magan Montoto, Y. Sugar Alcohols and Other Organic Compounds as Phase Change Materials; Vienna University of Technology: Vienna, Austria, 2018. [Google Scholar]
- Izutsu, K.; Koide, T.; Takata, N.; Ikeda, Y.; Ono, M. Characterization and Quality Control of Cocrystals. Chem. Pharm. Bull. 2016, 64, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, in Vitro and in Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino Acids as Co-Amorphous Stabilizers for Poorly Water-Soluble Drugs—Part 2: Molecular Interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Cao, F.; Da Rocha, A.B.D.O.; Cardoso, S.G.; Rodríguez-Hornedo, N. Cocrystals to Facilitate Delivery of Poorly Soluble Compounds Beyond-Rule-of-5 Graphical Abstract HHS Public Access. Adv. Drug Deliv. Rev. 2016, 101, 143–166. [Google Scholar] [CrossRef]
- Bavishi, D.D.; Borkhataria, C.H. Spring and Parachute: How Cocrystals Enhance Solubility. Prog. Cryst. Growth Charact. Mater. 2016, 62, 1–8. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical Cocrystals: A Review of Preparations, Physicochemical Properties and Applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Kiyonga, E.M.; Kekani, L.N.; Chidziwa, T.V.; Kahwenga, K.D.; Bronkhorst, E.; Milne, M.; Poka, M.S.; Mokhele, S.; Demana, P.H.; Witika, B.A. Nano- and Crystal Engineering Approaches in the Development of Therapeutic Agents for Neoplastic Diseases. Crystals 2022, 12, 926. [Google Scholar] [CrossRef]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical Cocrystals: New Solid Phase Modification Approaches for the Formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.H.; Augis, L.; Fourmentin, S.; Barratt, G.; Legrand, F.X. Deep Eutectic Solvents for Innovative Pharmaceutical Formulations. In Deep Eutectic Solvents for Medicine, Gas Solubilization and Extraction of Natural Substances; Fourmentin, S., Gomes, M.C., Lichtfouse, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 41–102. ISBN 9783540228608. [Google Scholar]
- Paiva, A.; Craveiro, R.; Aroso, I.; Martins, M.; Reis, R.L.; Duarte, A.R.C. Natural Deep Eutectic Solvents—Solvents for the 21st Century. ACS Sustain. Chem. Eng. 2014, 2, 1063–1071. [Google Scholar] [CrossRef]
- Vanda, H.; Verpoorte, R.; Klinkhamer, P.G.L.; Choi, Y.H. Natural Deep Eutectic Solvents: From Their Discovery to Their Applications. In Deep Eutectic Solvents: Synthesis, Properties, and Applications; Ramón, D.J., Guillena, G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 61–81. ISBN 9783527818471. [Google Scholar]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Solubility Advantage of Sulfanilamide and Sulfacetamide in Natural Deep Eutectic Systems: Experimental and Theoretical Investigations. Drug Dev. Ind. Pharm. 2019, 45, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Natural Deep Eutectic Solvents as Agents for Improving Solubility, Stability and Delivery of Curcumin. Pharm. Res. 2019, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural Deep Eutectic Solvents as New Potential Media for Green Technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef]