


1. Introduction
Obesity is an excessive fat deposition in the body, and it can be considered an independent multifactorial disease or a syndrome accompanying other conditions. In 1997, WHO proposed the most common classification of obesity based on body mass index (BMI (kg/m2)) [1]. Such a definition describes obesity as having a BMI more than or equal to 30, whereas overweight is defined as having a BMI larger than or equal to 25. The global prevalence of obesity and the burden of its comorbidities is increasing [2,3]. In 2016, according to WHO, over 650 million of the World’s adult population (11% of men and 15% of women) were obese [1].
Although obesity is a disorder that affects people of all ages, many epidemiological studies have found an increased risk of obesity from puberty until late middle age and a decrease after that [4,5,6,7,8]. Moreover, it has been established that obesity accelerates the aging process [9]. Multiple studies have demonstrated similar physiological mechanisms underlying obesity and aging [4,5,6,7,10,11,12,13,14,15,16,17,18,19,20,21]. The results of recent studies suggest that aging is the most substantial risk factor for developing metabolic syndrome and obesity [9,22]. Nevertheless, the influence of aging processes on the development of obesity is not well understood.
Age-related obesity is a complex process that depends on many factors, among which hormones such as leptin and insulin play a pivotal role [23]. Leptin regulates appetite, food intake, and energy balance by activating specific neuronal signals in the hypothalamus [24,25]. According to the early hypothesis, obesity occurs due to hypothalamic dysregulation of food intake and energy homeostasis caused by dysregulation of the adipokine leptin and decreased tissue sensitivity to its action, and hyperleptinemia and leptin resistance develop with increasing age [10,26,27,28,29,30,31,32]. Thus, leptin resistance may play a crucial role in age-associated weight gain and obesity, although underlying mechanisms are poorly understood.
In turn, hyperinsulinemia and insulin resistance are significant hallmarks of aging and obesity [33]. The relationships between insulin and its metabolic partner, leptin, play an essential role in energy homeostasis [34]. It has been shown that chronic insulin exposure can induce leptin secretion and production [35,36,37,38,39] and hyperinsulinemia causes leptin resistance in hippocampal neurons [34,40,41,42]. Vice versa, leptin can influence insulin and insulin resistance [43,44]. Although some researchers have not found an association between insulin and leptin levels [45,46], most in vitro, in vivo, and clinical studies indicate that there is a close relationship between these two hormones, and the role of leptin in age-related obesity is insulin-dependent [34,35,36,37,38,39,40,41,42]. Yet the interactions between these key regulators of energy metabolism are complex and poorly understood [47]. In this regard, the signaling pathways common to insulin and leptin, particularly the primary molecular players involved in the crosstalk between insulin, leptin, and other factors, require special attention.
Recent findings have pointed to the Rho GTPase Cdc42 as an integral part of regulating insulin secretion [48] and aging processes [49]. Cdc42 is a member of the Rho family of GTPases that regulates actin cytoskeleton, vesicle trafficking, cell polarity, morphology, proliferation, motility, and migration [50]. Upon activation, Cdc42 changes from an inactive GDP-bound form to an active GTP-bound state and back again, and these transformations are controlled by various proteins and regulatory factors [51]. The active form of Cdc42-GTP interacts with multiple effector proteins (P21-activated kinases, WASP and N-WASP, IQGAPs, MRCK, NADPH, Par proteins, etc.) and participates in signaling pathways that regulate growth, survival, differentiation, and many other aspects of cell biology [51]. Therefore, disturbances in the regulation of Cdc42 are implicated in many diseases, including cancer and neurodegenerative diseases [49,52].
Although the role of Cdc42 in glucose metabolism and interactions with insulin are relatively well understood, the results of the studies on Cdc42 and leptin interactions represent scattered data. Therefore, the review aims to summarize current knowledge on the role of Cdc42 in the crosstalk between the insulin and leptin pathways contributing to the development of age-related obesity. In addition, this article discusses the potential therapeutic implications of the Cdc42 pathway to mitigate obesity.
2. Age-Related Obesity and Insulin Imbalance
The relationship of human weight with age has two distinct patterns: an increase in the prevalence of obesity in middle age [53,54], and anorexia, weight loss, sarcopenia, and senile cachexia in the elderly after age 75 [55]. In general, people aged 75 years and older have a lower prevalence of obesity compared with 65–74 years old adults [56]. The results of multiple cross-sectional national surveys and a national longitudinal study of Australian women, which started in 1996 and involved more than 57,000 women in four age cohorts, have demonstrated that age-associated weight gain lasts from adolescence until late middle age [11]. Similar effects have been observed in nonhuman primates [12,13], dogs [14], cats [15], rats [16], and mice [17,18,19,20,21]. An Australian longitudinal study has also demonstrated that being overweight in middle age is strongly associated with a higher incidence of chronic diseases [57]. This finding agrees with a recent prospective cohort study in the US showing that overweighed middle-aged people have a higher burden of morbidity yet unaffected overall life expectancy. In comparison, in obese people, a higher burden of morbidity is accompanied by a shorter overall life expectancy [58]. In addition, modeling the trajectory of BMI over 28 years in women revealed an increased risk of dementia in obese women in their 50s but not in their 60s or 70s [59].
One of the reasons for the tendency of people in middle age to become overweight or obese is an age-related increase in total fat mass [60,61,62,63], which may also occur independently of gaining weight [64,65,66,67]. Total human fat mass consists mainly of two major compartments of white adipose tissue: subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT). Significant anatomical, cellular, molecular, physiologic, clinical, and prognostic differences exist in these adipose tissues in the body [68]. Numerous studies have demonstrated that VAT is a pro-inflammatory tissue with a higher number of inflammatory macrophages than SAT. It is associated with metaflammation (systemic and local inflammation), which can be quantified in organs [60,61,62,68]. In turn, with age, fat tends to accumulate predominantly in the abdominal and visceral areas and penetrate into muscles and bones [63,69], with noticeable gender differences in the dynamics of subcutaneous and visceral adipose tissue distribution. For instance, early studies have indicated that men reach their maximum subcutaneous fat volume at 40–50 years, while women up to 70 years [70]. Furthermore, in women, there is a sharp, almost four-fold increase in the amount of visceral fat between the ages of 25 and 65 years [71], while in men the mass of visceral adipose tissue increases only two-fold over the same period [72]. Although fat accumulation in adulthood is associated with many factors including decreased physical activity, high-calorie diet, stress, sleep patterns, etc., the crucial contributor to age-related obesity is a shift in hormonal balance and consequent reduction in the metabolic rate [73,74,75]. Extensive research and clinical studies have shown that age-related complex changes in the endocrine system affect the secretory pattern of hormones, tissue sensitivity to hormones, and imbalance of hormone levels [76,77,78].
The expansion of total fat mass depends on adipocyte proliferation (hyperplasia) and an enlargement of adipocyte size (hypertrophy) [79]. The results of several studies have found that there is a balance between hyperplasia and hypertrophy in different adipose tissue depots [64,65]. In general, hypertrophy precedes hyperplasia, and when adipocytes reach the lipid accumulation limit, adipocyte proliferation and/or differentiation are triggered [66,67]. An imbalance between hyperplasia and hypertrophy may exacerbate the metabolic outcome of obesity [80,81,82]. Insulin is one of the main endocrine hormones regulating energy and lipid metabolism, and it is actively involved in stimulating both hypertrophy and hyperplasia of adipocytes [83,84,85]. Exposure to insulin leads to the elevation of the absorption of glucose, as well as the storage of free fatty acids and triglycerides in adipocytes [86]. In addition, insulin suppresses lipolysis and promotes de novo synthesis of fatty acids in adipose tissue cells [87]. Insulin is vital to the late stages of adipogenesis because it enhances gene expression of various fat-specific transcription factors and is required to achieve a fully functional adipocyte phenotype [88,89,90].
Gender differences in age-related obesity between men and women may also originate from the differences in insulin secretion and tissue sensitivity to insulin [91,92]. Glucose-stimulated insulin secretion patterns may vary, with women exhibiting a higher level of secretion [93]. Studies show that women are generally more insulin-sensitive than men, while males have more insulin and high-affinity receptors [94,95,96]. As individuals age, both genders experience a decrease in peripheral insulin sensitivity [96,97]. Sex hormones positively affect insulin, so while testosterone sensitizes tissues to insulin [98], estrogens, in addition to increasing tissue sensitivity to insulin, also stimulate the synthesis and release of insulin by the pancreas [99]. Insulin resistance impacts both men and women, with a higher prevalence observed in men. However, following menopause, the incidence in women rises and becomes comparable to that in men. Changes occurring postmenopausal may heighten the risk of insulin resistance and the development of type 2 diabetes [100].
Hyperinsulinemia, also known as dysregulated insulin production and/or clearance, is a prevalent feature of obesity and metabolic diseases that results in persistently increased insulin levels without hypoglycemia [101,102]. Hyperinsulinemia is multifactorial and related to a number of causes, including genetic predisposition, lifestyle, diet, and environment [101]. Several epidemiological and laboratory studies have shown a complex relationship between age and insulin secretion and clearance: while increased insulin secretion and hyperinsulinemia are more prominent in late middle age, decreased insulin secretion is observed in advanced old age [103,104,105,106,107,108]. On the other hand, the data about the effect of aging on insulin clearance are contradictory: some studies find no difference [105,109], while others report a decreased insulin clearance in aged rodents and humans [110,111,112]. Kurauti et al. hypothesized that insulin clearance is not impaired at younger ages due to compensatory activation of insulin-degrading enzyme (IDE) expression. However, at later ages, this compensatory mechanism breaks down, leading to decreased insulin clearance and increased hyperinsulinemia [103,105].
Aging and obesity cause immune dysregulation in adipose tissue, leading to chronic inflammation with increased infiltration and activation of innate and adaptive immune cells [113,114,115,116]. The number of macrophages, the primary adipose tissue immune cell population, increases with fat mass accumulation and aging, potentially accounting for up to 40% of all adipose tissue cells [117,118,119]. Adipose tissue chronic inflammation is triggered by the secretion of several inflammatory factors and an increase in pro-inflammatory macrophages in the adipose tissue associated with obesity [120,121]. Although the link between insulin resistance and inflammation remains unclear, data demonstrate that inflammation can lead to the development of insulin resistance [122]. Furthermore, it has been shown that proinflammatory cytokines can also stimulate insulin production [123]. In turn, hyperinsulinemia and insulin resistance may promote myelinogenesis and common myeloid progenitor (CMP) granulocyte/monocyte progenitor (GMP) proliferation through the FoxO signaling pathway [124]. Yet it has recently been shown that insulin resistance triggered by obesity emerges before the accumulation of macrophages and the onset of inflammation in adipose tissue [125].
3. Age-Related Obesity and Leptin Resistance
The age-related increase in fat mass and hyperinsulinemia triggers an imbalance of leptin-dependent regulation of adipose tissue homeostasis [126]. Leptin is a member of a family of long-chain class-I helical cytokines that includes the IL-6, G-CSF, erythropoietin, thrombopoietin, growth hormone, and prolactin [127]. Leptin is found in all mammals and is characterized by a highly preserved primary amino acid sequence and its functions [128]. The human LEP gene is located on chromosome 7 and encodes a 167 amino acid peptide with a molecular weight of 16 kD [129,130]. Leptin is produced mainly by adipocytes and is considered a crucial adipostatic factor. Under normal physiologic conditions, high leptin levels decrease adipose tissue mass by activating hypothalamic control of energy expenditure, thermogenesis, and calorie intake [25,131,132]. Therefore, serum levels of leptin are proportional to the amount of energy stored in adipose tissue and fluctuate with significant changes in calorie intake [133,134]. However, in obesity and aging, leptin malfunction is associated with decreased sensitivity of tissues to leptin [25].
Leptins bind to the obesity receptor (Ob-R). There are six isoforms of Ob-R divided into three classes (long, short, and secretory isoforms), which are the products of alternative RNA splicing of the db gene. A long, fully active isoform of Ob-Rb is expressed mainly in the hypothalamus, where it takes part in energy homeostasis and in the regulation of secretory organ activity. Ob-Rb is also present in all types of immune cells and is involved in innate and adaptive immunity. Short leptin isoforms co-called Ob-Ra, Ob-Rc, and Ob-Rd are able to bind JAK kinases and activate some signal transduction cascades. A soluble isoform (Ob-Re) can regulate serum leptin concentration and serve as a carrier protein delivering the hormone to its membrane receptors and is able to transduce the signal into the cell [135]. Gancarz et al. found that aging decreases the soluble form of Ob-Re and Ob-Ra in human monocytes, while the long form of the leptin receptor Ob-Rb remains unchanged [136].
Over the past 20 years, the classic hypothalamic leptin–melanocortin model of leptin’s anorexigenic effects has been proposed [137,138]. Within this model, leptin is thought to bind to its receptor, the so-called long form of the leptin receptor (Ob-Rb), which is expressed by two antagonistic populations of neurons in the arcuate nucleus of the hypothalamus (ARC): proopiomelanocortin (POMC)-containing neurons and neuropeptide Y (NPY)/agouti-related peptide (AgRP) (AGRP/NPY) neurons [139,140]. Leptin binding to Ob-Rb leads to activation of POMC neurons, which produce anorexigenic molecules such as αMSH (α-melanocyte-stimulating hormone) and deactivation of AGRP/NPY neurons. This results in decreased food intake and energy expenditure. At low leptin levels, there is deactivation of POMC neurons and activation of AGRP/NPY neurons, which produce AGRP/NPY orexigenic peptides, resulting in increased appetite. However, recent studies demonstrated that this model is insufficient for understanding leptin regulation of energy homeostasis [137,141]. A line of evidence has shown that leptin’s effects on eating behavior are not due to direct effects on POMC neurons [137,142,143]. While the direct action of leptin on POMC neurons is more likely related to the maintenance of normal glucose homeostasis [142,144], it is hypothesized that the specific effects of leptin are mediated by multiple populations of leptin-sensitive non-POMC neurons [137,145,146,147,148]. Thus, the current understanding of leptin’s regulation of hypothalamic neuronal circuits that control feeding and energy expenditure is incomplete and needs to be revised.
After discovering leptin in 1994, it was hypothesized that increased leptin concentrations should have led to decreased appetite and increased energy expenditure in obesity [25]. However, treatment with leptin in obesity did not produce the expected effects. To explain this paradox, the concept of leptin resistance has been proposed by analogy with insulin resistance [30,133]. Leptin resistance is described as a diminished sensitivity or lack of response to endogenous or exogenous leptin. It is characterized by high circulating leptin concentrations (hyperleptinemia) and a decreased tissue sensitivity to leptin [28,30,149]. Traditionally, two main types of leptin resistance are distinguished: reduced tissue sensitivity to leptin in the brain (central leptin resistance) and in the peripheral tissues (peripheral leptin resistance) [150]. The development of leptin resistance requires a combination of high endogenous leptin levels in the blood and a high-fat diet; separately, these factors cannot lead to the development of leptin resistance [151].
A number of studies indicate that aging is a risk factor for the development of leptin resistance and obesity. However, the causative relations between leptin resistance and age-related obesity are disputable [28,29,152,153,154,155]. Some authors believe leptin resistance develops in response to obesity, while some findings suggest that obesity develops in response to aging-associated leptin resistance. For instance, a recent study found that both male and female Wistar rats showed an age-related increase in plasma leptin concentrations [156]. Transgenic mice with chronically elevated leptin levels exhibited a lean phenotype at a young age, whereas at 33–37 weeks, there was an increase in body weight, significant accumulation of fat mass, and lipid deposition in adipocytes [157]. Studies in male F-344 × BN rats have shown that leptin gene expression in inguinal white adipose tissue and circulating leptin levels increase with age without increasing adiposity [16]. In contrast, in C57BL/6J mice, both serum leptin concentrations and leptin gene expression levels decreased with age, while body weight remained stable [17]. Similarly, Ma et al. showed that basal leptin levels in the blood of F-344 × BN rats were two-fold higher in 4-month-old F344 × BN rats compared with 21-month-old rats. At the same time, an exogenous increase in plasma leptin levels resulted in a 50% suppression of leptin gene expression in younger animals. Still, it did not inhibit leptin gene expression in aging rats [27]. Yet these conflicting results may be due to differences in study design based on normalization of leptin levels [158]. Moreover, several lines of evidence suggest that age-related changes in leptin levels in humans differ significantly from their rodent counterparts [29,158].
It is worth mentioning that men and women differ in the leptin levels they synthesize and secrete and in their responses to endogenous and exogenous leptin [159,160]. Several studies indicate that women have higher plasma leptin levels than men, regardless of the variation in total body fat mass [159,161]. The predominance of subcutaneous adipose tissue in women, which is distinguished not only by large mass but also by greater expression and secretion of leptin compared with men, is thought to be the source of sexual dimorphism in leptin levels [159,161,162]. Estrogens enhance leptin sensitivity [163], while androgens induce leptin resistance and dysfunction [164,165,166]. It has been found that estradiol and estradiol receptor levels drop with age, especially during menopause [167]. It is hypothesized that the age-related decrease in estradiol receptor expression reduces the effect of this hormone on leptin [168]. Testosterone is also likely to play a crucial role in sexual dimorphism, reducing the growth of subcutaneous fat tissue, leptin secretion, and gene expression [164,165,166]. Males and females respond differently to leptin, with female brains being more sensitive to it [169]. Estradiol may enhance hypothalamic expression of the long form of the leptin receptor, potentially modulating central leptin sensitivity in female gonadal hormones [170]. When exposed to a high-fat diet, males develop leptin resistance earlier than females [171,172,173].
The causes of a decrease in tissue leptin sensitivity can generally be classified into two mechanisms: the first is linked to impaired transport of leptin to its receptors, while the second includes mechanisms leading to a decrease in signal transmission from the receptor to the downstream effectors [149,174]. Leptin is subsequently released into the bloodstream, where it can exist in a free active form and an inactive form bound to leptin-binding proteins Ob-Re [175,176,177,178]. The ratio of a free form of leptin to Ob-Re is an important indicator of leptin bioavailability called the free leptin index (FLI = leptin/Ob-Re) [179]. The concentration of human OB-Re in serum is proportional to the amount of membrane-bound leptin receptor [180]. It is formed by proteolytic cleavage of the membrane-bound leptin receptor by metalloproteases, mainly in the liver [181].
Obesity leads to a decrease in Ob-Re levels and an increase in FLI [178,182,183,184]. In turn, decreased blood Ob-Re levels are associated with leptin resistance linked to obesity and diabetes [185]. Lean individuals have more bound leptin in their blood, and weight loss is associated with increased levels of Ob-Re in the blood of both humans and rodents. It is hypothesized that decreased Ob-Re levels in obese and diabetic patients lead to increased leptin clearance and may enhance leptin-induced leptin resistance [181]. Some data suggest that an imbalance between the free form of leptin and Ob-Re may be one of the most important mechanisms for the development of leptin resistance [47,186]. Several studies have also shown that FLI seems to change with age [136,187,188]. For instance, a 2015 study found that the median FLI was substantially greater in healthy elderly, non-obese males (aged 64.7 ± 3.1 years) than in young males (aged 26.8 ± 3.6 years) [136]. Leptin-binding activity levels were shown to be low at birth, high in the years before puberty, reduced through puberty, and then remained steady throughout adulthood [188].
A likely reason for the drop in Ob-Re levels in the blood during obesity and aging may be a combination of chronic hyperleptinemia and endoplasmic reticulum stress (ER stress) in hepatocytes [189]. Exposure to leptin can lead to temporary ligand-induced suppression of the receptor [190]. Under normal conditions, the membrane pool of the leptin receptor is replenished. Still, with obesity and aging, misfolded proteins accumulate in the hepatocytes’ endoplasmic reticulum, the so-called ER stress, which slows the return of leptin receptors to the plasma membrane [189]. A recent study suggests that activation of the endocannabinoid CB1R system in the liver, which induces ER stress and suppresses CHOP protein, may reduce Ob-Re expression in hepatocytes [186]. The CB1R system, linked to metabolic syndrome, including leptin and insulin resistance, is triggered by both central and peripheral stimulations during obesity [191,192]. Aging leads to increased metabolic signatures of chronic ER stress, and in the liver, in particular, decreased ER stress with age may contribute to metabolic diseases [193].
In turn, multiple mechanisms of energy homeostasis regulation are triggered by binding leptin to its membrane-bound receptor (Ob-Rb) expressed in leptin-sensitive neuronal populations [194,195]. There is an assumption that the reduced expression of Ob-Rb in the brain and peripheral tissues is one of the crucial reasons for leptin resistance [196]. In rodents, age-related decline in leptin receptor expression was detected in the brain, indicating a relationship between aging and leptin resistance. For instance, it was reported that the amount of leptin receptor protein in the hypothalamus of aged obese rats was reduced by half compared with young animals [196]. A study by Fernández-Galaz et al. showed that older rats had lower levels of Ob-Rb mRNA in their hypothalamus in comparison with the young rats, but food restriction restored this parameter back to the levels seen in young rats [197]. Similar findings have been reported in another study, where Ob-Rb expression was markedly reduced in the brains of aged 5XFAD mice [198]. Guadalupe-Grau and coauthors found that leptin receptor expression was lower in vastus lateralis muscle biopsies from aged men (58 ± 8 years) in comparison with young men (24 ± 4 years) groups [199].
It is suggested that leptin receptor degradation is another mechanism of leptin resistance in obesity and aging, and it could be associated with increased activity of the matrix metalloproteinase-2 (Mmp-2) [200]. It has been found that HFD-induced obesity stimulated Mmp-2 protein activation within the hypothalamus with subsequent cleavage of the extracellular domain of the leptin receptor [200]. Mmp-2 is a member of a family of matrix metalloproteinase (MMP) involved in the resorption of extracellular matrix and other processes contributing to age-related diseases [201,202,203]. Mmp-2 activity increases with age in various tissues, including blood vessels, and perhaps this age-related change in Mmp-2 protein activity translates into age-related leptin receptor status [203]. In addition, physiologically elevated insulin levels can induce a dramatic increase in Mmp-2 activation in blood vessels [204].
4. Cdc42, Obesity, and Aging
Aging may be associated with both increased and decreased Cdc42 activity and expression, which varies for different cells and tissues. For instance, Cdc42 activity is significantly increased in vessels, adipose tissue, liver, blood cells, and kidney [49], while the downregulation of Cdc425 is observed only in the brain and some other organs during the aging process [49]. Notably, Cdc42 activity changes with age in metabolism-related organs expressing the leptin receptors, such as brain [205,206,207,208], adipose tissue [209,210], liver [206], and kidney [208,211]. Li et al. demonstrated that the hippocampus of old rats had reduced activity of Cdc42 compared with young animals, but 12 weeks of aerobic exercise significantly increased the activity of Cdc42 [205]. In contrast to the brain, Cdc42 activity increases with age in adipose tissue [209,210]. One reason for the increase in Cdc42 activity may be related to the change in adipocyte size, which is affected by age and obesity: average adipocyte size increases in middle and old age and then decreases over time [212]. Changes in adipocyte size are also associated with the reorganization of the actin cytoskeleton [213]. Although the study on the impact of obesity on Cdc42 activity and expression is limited, there are data demonstrating that after two weeks of a high-fat diet (HFD), C57BL6/J mice exhibited dramatic remodeling of the actin cytoskeleton and increased Cdc42 activity along with increased cell size and impaired insulin signal transduction in adipocytes [214]. Moreover, the activation of Cdc42 has been found to be increased in CD4+ T cells in obese children [215].
A mouse transcriptome study showed that obesity modulates Cdc42 expression in different mouse organs in an age-dependent manner [216]. Thus, microarray data obtained from 4-week- and 10-week-old lean and obese C57BL/6 mice and BTBR mice (mice line with ob/ob leptin-deficiency mutation) (GEO accession: 10785) demonstrated that obesity leads to a dramatic increase in Cdc42 expression in adipose tissue of C57BL/6 mice regardless of age, and a less significant upregulation of Cdc42 mRNA in BTBR mice, which was more robust in older animals. Increased expression of Cdc42 was also observed in obese livers, with the most significant changes observed in the obese C57BL/6 mice group at ten weeks of age, whereas in mice of the same line at four weeks of age, Cdc42 levels were unchanged in obesity. In the BTBR mice groups, the induction of Cdc42 in the liver during obesity was independent of age but was less pronounced than in C57BL/6 mice. In the pancreas of C57BL/6 mice, obesity caused a decrease in Cdc42 expression, especially in four-week-old animals, whereas older animals showed a slight reduction. Similar dynamics were observed in BTBR mice, where age also leveled out the decrease in Cdc42 expression. In the hypothalamus of mice, obesity and age did not lead to changes in Cdc42 expression. This aligns with recent findings indicating altered Cdc42 activity in the hypothalamus of aged rats, while the overall expression level of Cdc42 remained unchanged compared with their younger counterparts [205]. Thus, it is evident that obesity mostly has an increased impact on Cdc42’s ability to operate in a variety of tissues.
Cdc42 is a vital protein in angiogenesis and vasculogenesis as well, and its dysfunction can potentially lead to vascular dysfunction [217]. Mammoto et al.’s study revealed that Cdc42 activity in endotheliocytes from adipose tissue in individuals over 50 significantly increased compared with younger individuals [218]. The study shows that increased Cdc42 activity leads to an age-related increase in endotheliocyte size and senescence, decreased cell proliferation, and angiogenesis through aberrant Cdc42 -YAP1 signaling [218]. Moreover, a study by Ito et al. demonstrated that Cdc42 mediates p53-induced vascular inflammation in vivo, increases the expression of proinflammatory molecules in endothelial cells by activating the NF-κB pathway, and plays a crucial role in endothelial Cdc42 in chronic inflammation and the development of atherosclerosis [219]. Besides its impact on endothelial cells, Cdc42 regulates crucial events in adventitial progenitor cells and vascular smooth muscle cells [220,221,222]. Cdc42 controls the migration of Sca-1+ adventitial progenitor cells, leading to the accumulation of VSMC during vascular wall remodeling [220,222]. The activation of Cdc42 by protein kinase C delta type initiates the production of type I collagen from vascular smooth muscle cells [221]. Aging increases Cdc42 signaling in VSMC [223], potentially increasing collagen I secretion and leading to vascular fibrosis and atherosclerosis complications [224]. Moreover, overexpression of Cdc42 may increase arterial stiffness due to enhanced osteogenic differentiation of VSMC and subsequent vascular calcification [225]. Even though the precise relationship between Cdc42, insulin, and leptin in blood vessels is unknown, it is reasonable to assume that Cdc42 activation is linked to vascular remodeling caused by age-related hyperinsulinemia and hyperleptinemia. Aging exacerbates obesity-induced vascular pathology, whereas activation of Cdc42 regulates vascular function positively in young organisms [226,227].
Although gender changes in Cdc42 expression and activity are not well understood, limited research suggests that sex hormone influences are closely related to Cdc42 activity [228,229,230,231]. For instance, it has been shown that estrogens inhibit the activity of Cdc42 [230], and androgens, on the contrary, activate Cdc42 [231]. Additionally, Cdc42 has been demonstrated to inhibit the transcriptional activity of estrogen receptor alpha [228]. Clinical studies also partially support the connection between Cdc42 and estrogens: a recent study found increased expression of the Cdc42 gene in saliva associated with menopausal status, with the most significant increase observed in women over 45 years of age [229]. These observations may suggest that age-related declines in estrogen and androgen levels may influence Cdc42 regulatory pathways with subsequent changes in cellular processes and energy homeostasis.
comprehensively outlines the multifaceted impact of age-induced changes in Cdc42 activity on the development and progression of obesity.
5. Crosstalk between Cdc42 and Adipoinsular Axis
As mentioned earlier, Cdc42 is a Rho GTPase protein that plays a vital role in the regulation of many cellular functions [50] and metabolic processes [238]. Several studies have identified a novel role for Cdc42 in maintaining glucose metabolism and controlling blood glucose levels through the regulation of cellular processes in metabolically active tissues such as skeletal muscle and adipose tissue, as well as the pancreas [48]. Cdc42 activates the second phase of glucose-induced insulin secretion in pancreatic islets by regulating granule fusion, exocytosis, and actin cytoskeleton rearrangement [239,240]. In addition, it is hypothesized that Cdc42 may influence pancreatic β-cell proliferation through the activation of effectors like PAK1 and CyclinD1 [48]. Furthermore, in 3T3-L1 adipocytes, Cdc42 activation mediates insulin-stimulated GLUT4 translocation and glucose transport in a PI3-kinase-dependent manner [241]. In turn, insulin treatment can enhance the Cdc42 activity and the association of Cdc42 with activated PI3-kinase (PI3K) in these cells [241]. It is worth mentioning here that PI3K plays an important role in the body’s energy balance in a variety of organs [242], and the PI3K/AKT pathway is critical for energy metabolism in insulin-sensitive tissues [242].
Veluthakal et al. demonstrated that despite the same expression level of Cdc42 in islets of people with and without type 2 diabetes (T2DM), glucose barely activated Cdc42 in islets of people with T2DM [243]. It was established that patients with T2DM have an 80% loss of expression of the critical effector of Cdc42, PAK-1, involved in the regulation of insulin secretion in comparison with healthy people [244]. In addition, a comprehensive bioinformatic analysis of 981 genes revealed that Cdc42 could potentially be used as a candidate gene target for diagnosing and treating this disease [245]. The latest research on the effect of Cdc42 deletion in pancreatic β-cells and hypothalamus showed that Rip-CDC42cKO mice exhibited glucose intolerance, a significant decrease in glucose-induced insulin secretion in isolated islets, and decreased insulin sensitivity in peripheral tissues [141]. The possible role of Cdc42 in the development of peripheral insulin resistance is evidenced by studies that have shown altered activity of Cdc42-related insulin signal transduction factors such as Cdc42 interacting protein-4 (CIP4) and G protein-coupled receptor kinase 2 (GRK2) in adipose tissue [211,246] and C9orf72 in the liver [247].
Change in lipid metabolism caused by obesity and aging is a condition known as atherogenic dyslipidemia. This condition is characterized by increased levels of triglycerides (TG) and/or low-density lipoprotein (LDL) and decreased levels of high-density lipoprotein (HDL) [248,249,250]. Epidemiological studies consistently show that atherogenic dyslipidemia is a significant risk factor for the development of atherosclerosis and metabolic disorders [101,102,251,252,253]. In turn, disruption of insulin signaling may be one of the causes of atherogenic dyslipidemia in aging and obesity [254]. Hyperinsulinemia stimulates the production of VLDL triglycerides [255,256], and elevated insulin secretion raises the concentration of low-density lipoprotein cholesterol [257].
Insulin resistance, which is inversely correlated with HDL and positively correlated with TG and LDL, is another critical factor that significantly contributes to the development of atherogenic dyslipidemia. [252,256]. Insulin resistance is closely linked to obesity, forming a dynamic interaction that has long intrigued researchers, and the intricate cause-and-effect relationship between insulin and insulin resistance remains a subject of ongoing debate [258]. However, recent findings suggest that hyperinsulinemia may precede the onset of insulin resistance [102,259]. Studies using mutant mice with impaired insulin clearance revealed impaired insulin clearance and hyperinsulinemia at 2 months, followed by hepatic insulin resistance at 6–7 months. This was subsequently accompanied by visceral obesity, hyperphagia, hyperleptinemia, and hypothalamic leptin resistance [260].
An age-dependent [248] or obesity-induced [261,262] increase in free fatty acid (FFA) levels is considered a likely mechanism causing changes in lipid metabolism through impaired insulin signaling. It has been demonstrated that sharply increasing FFA can reduce insulin action [263] and cause adipose tissue to express and secrete proinflammatory cytokines, which, in turn, reduce insulin sensitivity [264]. Einstein et al. demonstrated that the inflammatory response to nutrient excess and susceptibility to FFA-induced insulin resistance are exacerbated by aging [265]. One of the major signaling pathways involved in the development of FFA-induced liver insulin resistance is the c-Jun N-terminal kinase (JNK) pathway, which contributes to the disruption of insulin signaling and promotes cell damage (lipoapoptosis) [266,267]. In turn, Cdc42 is involved in the regulation of liver lipid status as a major contributor to the saturated fatty acid-stimulated JNK pathway in hepatocytes, which promotes lipoapoptosis and NAFLD [268]. In addition, Cdc42 activation leads to increased fluid-phase pinocytosis of LDL by macrophages [234,235,236], causing them to absorb excess lipids and turn into foam cells [269], thereby enhancing in vivo atherosclerosis [236]. Therefore, age-related activation of Cdc42 in the liver and macrophages may contribute to dyslipidemia and atherosclerosis.
On the other hand, hyperleptinemia is a significant contributor to obesity and the disruption of energy homeostasis [270,271,272]. Under physiological conditions, leptin enhances insulin sensitivity by increasing fatty acid oxidation and glucose uptake through AMPK and PPAR-alpha activation [273]. Hyperleptinemia leads to an increase in insulin resistance and disruption of glucose metabolism [270,271,272]. A partial decrease in plasma leptin levels in obese patients can restore hypothalamic leptin sensitivity, reduce weight gain, and improve insulin sensitivity [271]. Upregulation of Cdc42 in obesity contributes to the development of insulin resistance through increased leptin production by hypertrophied adipocytes. Moreover, the possible role of Cdc42 in the development of peripheral insulin resistance is evidenced by studies that have shown altered activity of Cdc42-related insulin signal transduction factors such as Cdc42 interacting protein-4 (CIP4) and G protein-coupled receptor kinase 2 (GRK2) in adipose tissue [211,246] and C9orf72 in the liver [247].
Considering the close relationships between obesity, leptin resistance, insulin resistance, and aging, it is conceivable that Cdc42 dysfunction may be associated with leptin resistance [270,274]. Leptin has many different influences on Cdc42 activity depending on the type of cells it acts upon. Several studies described interactions with leptin and Cdc42 during neurogenesis in the hippocampus, in which leptin exposure activated Cdc42 in hippocampal neurons [275,276,277]. It has been reported that exposure of hippocampal neurons to leptin triggers a CaMKK/CaMKI signaling cascade that leads to phosphorylation and activation of β1Pix, a Cdc42/Rac guanine nucleotide exchange factor (GEF) [275]. It was also found that the leptin receptor and β-PIX form a complex, the amount of which temporarily increases upon activation of the leptin receptor during GABAergic synaptogenesis in hippocampal neurons [276]. In another study, leptin was shown to suppress the expression of p250GAP, a negative regulator of Rac, RhoA, and Cdc42, and this suppression is mandatory for enhancing synaptogenesis in hippocampal neurons [277].
The interaction between leptin and Cdc42 occurs not only in the brain but also in peripheral organs. Leptin exposure activates Cdc42 in mouse Sca-1+ vascular progenitor cells [222] and human brain microvascular pericytes [227] and causes enhanced angiogenesis and cell migration. The identification of mRNAs for differentially expressed genes after leptin treatment of ob/ob mice showed a 1.5-fold increase in Cdc42 expression in the liver [278]. In contrast, in rat cardiomyocytes, leptin did not affect Cdc42 activity [279]. In some studies, leptin induced PI3K-dependent reorganization of the actin cytoskeleton, leading to the depolymerization of F-actin in pancreatic β-cells of mice and rats [280,281]. Ning et al. discovered that leptin-induced F-actin depolymerization is linked to PTEN inhibition [281], which can activate Cdc42, causing actin cytoskeleton reorganization [136]. If considering the PI3K signaling in the brain, it is shown that it has a central role in the regulation of satiety by leptin [282]. In this regard, inhibiting PI3K by LY294003 abrogated the reduction in food intake stimulation by leptin [283]. In the ARC, leptin activates PI3K in the POMC neurons while indirectly inhibiting PI3K in the NPY/AgRP neurons [284], leading to suppression of food intake. In vivo, studies with neuron-specific PI3K ablation have pointed to a role of PI3K in leptin-mediated feeding behavior and body weight regulation. Mice with POMC-specific depletion of PI3K regulatory subunits p85α and p85β have normal food intake and body weight but fail to suppress food intake upon acute leptin administration [282]. Considering the fact that PI3K binds to Cdc42 via the N terminal region of the p85α regulatory subunit [241], it is suggested that both proteins may affect energy metabolism via a common signaling pathway.
In turn, Cdc42 may influence leptin secretion by mediating insulin action. Numerous studies have found insulin-induced long-term leptin secretion by fat cells through a transcriptional or post-transcriptional mechanism [35,285,286,287]. Zeigerer et al. demonstrated that treatment of 3T3-L1 adipocytes with insulin caused a two-fold increase in leptin secretion and could be blocked by brefeldin A [287]. Since mammalian Cdc42 is a Brefeldin A-sensitive component of the Golgi apparatus, it can be assumed that Cdc42 is involved in leptin secretion [288].
As indicated in the previous section, one of the pivotal regulators of leptin signaling is metalloproteinase MMP-2, the activity of which leads to cleavage of the extracellular domain of the leptin receptor in the hypothalamus [200]. Cdc42 can regulate VEGF-dependent MMP-2 activation in endothelial cells, and overexpression of Cdc42 enhances MMP-2 activity and capillary sprouting [289,290]. Since increased Cdc42 and MMP-2 activity are observed in obesity and aging, it is conceivable that increased Cdc42 activity may lead to the degradation of the leptin receptor and affect leptin transport to the brain.
As discussed in the previous chapter, ER stress plays a critical role in the release of the soluble form of the leptin receptor by the liver and the development of leptin resistance [189]. Obesity can lead to the development of ER stress through the suppression of hepatic autophagy [291]. N-WASP, expressed in hepatocytes, is activated by Cdc42, leading to actin polymerization and inhibiting autophagy. A recent study showed that increased Cdc42 activity in hepatocytes associated with insulin resistance leads to the suppression of lipophagy, a type of autophagy [247]. Several lines of evidence suggest that Cdc42 levels in the liver increase with aging [206,216,232]. Therefore, it can be assumed that increased age-related activity of Cdc42 in the liver may enhance leptin resistance through a decrease in soluble forms of the leptin receptor. Moreover, Cdc42 may have indirect effects on the development of leptin resistance through the regulation of leptin clearance. The main organ involved in leptin excretion is the kidneys. It excretes up to 80% of leptin, and renal dysfunction may actively contribute to hyperleptinemia [292]. Therefore, an indirect effect of Cdc42 on leptin levels is possible through renal dysfunction with Cdc42 activation in podocytes and renal vascular cells caused by hyperglycemia [225,293,294,295]. below summarizes the relationship between Cdc42 and the adipoinsular axis. We suggest that Cdc42 is an important element of the adipoinsular axis and age-related dysregulation of Cdc42 may exacerbate metabolic disturbances and lead to age-related obesity (Figure 1).
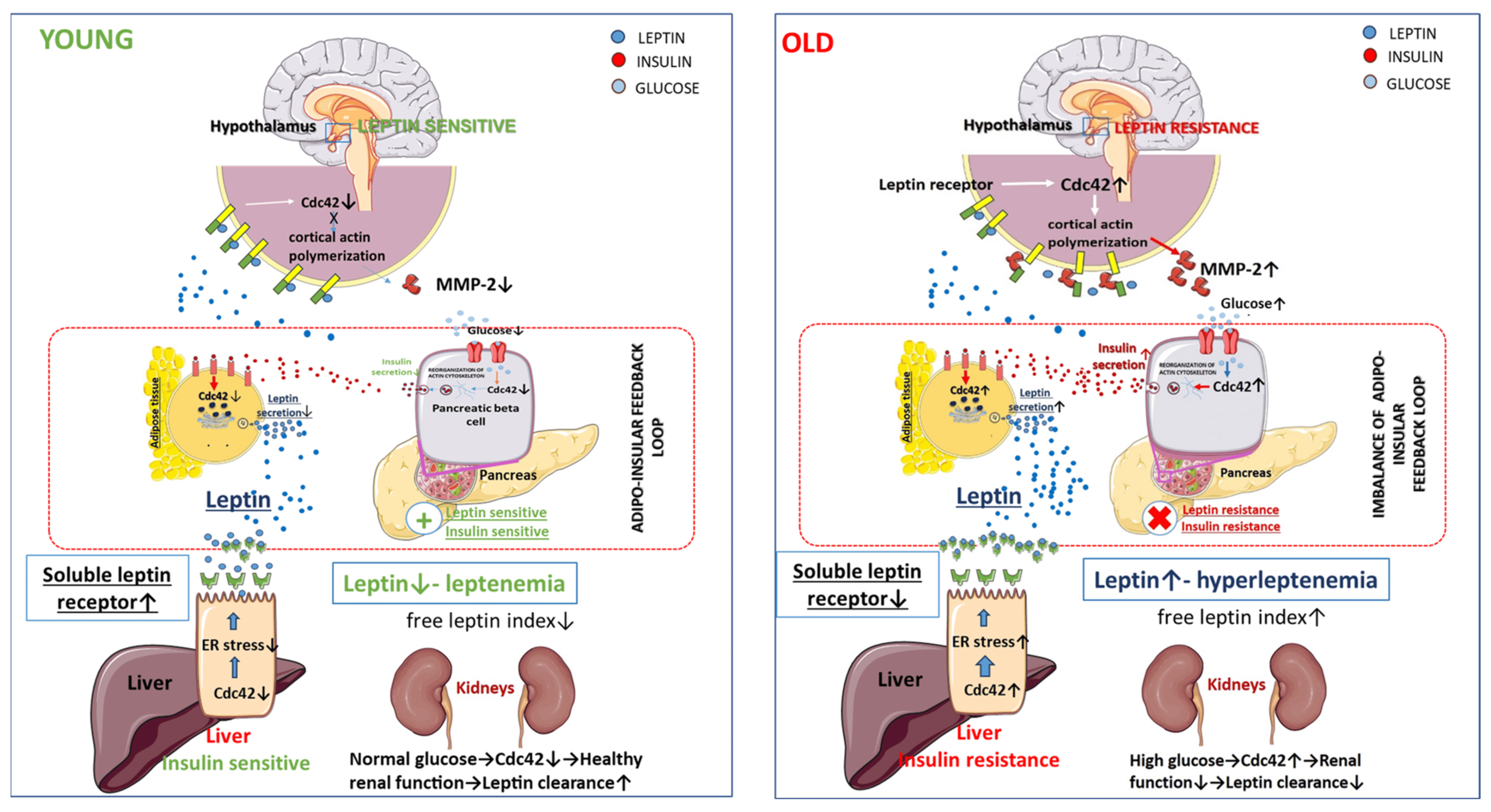
Figure 1. Role of Cdc42 in the imbalance of adipoinsular axis.
6. The Potential Therapeutic Implications of Cdc42 and Adipoinsular Axis to Mitigate Obesity
It is noteworthy to mention that some drugs used to sensitize tissues to leptin and affect obesity can modulate Cdc42 activity. Thus, the antidiabetic drug metformin, which has been reported to be able to reduce leptin and insulin levels and increase the expression of the leptin receptor gene (Ob-Rb) in the arcuate nucleus [313], has an inhibitory effect on Cdc42 [314]. Moreover, curcumin, a lipophilic polyphenol derived from the spice turmeric, which reduces weight, leptin, and leptin levels in patients with metabolic syndrome and related disorders [315,316], can inhibit Cdc42 activity [317].
Several clinical studies have shown that the polyphenolic compound with pleiotropic activity, resveratrol (trans-3,5,4′-trihydroxystilbene), can be used as a therapeutic strategy in the control of obesity [318]. The exact mechanisms of resveratrol’s effects on adipose tissue and glucose metabolism are not known, but it is interesting that resveratrol suppresses glucose-induced vascular smooth muscle cell migration via inhibition of Cdc42 [319]. However, these agents affect a wide range of signaling pathways, and it is difficult to assess the contribution of Cdc42 inhibition to their metabolic effects.
Long-chain omega-3 polyunsaturated fatty acids (n-3 PUFAs) have anti-inflammatory and hypotriglyceridemic properties, so the use of a diet based on them is being considered for the treatment and prevention of obesity and related diseases [320]. It has been shown that a diet rich in n-3 PUFAs improves insulin sensitivity and may prevent the development of insulin resistance in response to high-fat feeding and modulates the expression and secretion of adipocytokines [321,322]. Moreover, a diet based on n-3 PUFAs has a major impact on influencing leptin in the context of inflammation in obesity [323]. In addition, a 12-week open-label intervention study showed that consumption of 2.7 g/day of omega-3 polyunsaturated fatty acids led to a decrease in the level of Cdc42 mRNA in the blood of men [324]. In vitro and in vivo studies revealed that diets enriched with omega-3 polyunsaturated fatty acids suppressed Cdc42 activity in mice colonocytes [325].
These data suggest that nutritional interference of Cdc42 is possible, but the effects of different nutrients on Cdc42 regulatory dynamics are not fully understood and require further study. Furthermore, dysregulation of Cdc42, triggered by dietary factors or aging, may contribute significantly to the development of insulin and leptin resistance, highlighting its central importance in metabolic health. Although we have not found any studies on the effect of selective Cdc42 inhibitors on obesity, it has been shown that systemic administration of a selective small molecule, the Cdc42 inhibitor CASIN, can significantly reduce inflammatory processes in the body and increase life expectancy [237]. Therefore, it could be of great interest to evaluate the effects of Cdc42 inhibitors and activators on leptin resistance and obesity.
7. Conclusions
Obesity in middle age has profound implications, hastening the onset of chronic diseases and disability. Central to this process is the disruption of the adipoinsular axis due to aging, a crucial factor contributing to obesity. Although leptin and insulin are the key players that regulate adipose tissue homeostasis and energy metabolism, the complex interplay between these two hormones is still poorly understood, highlighting the need for the identification of new molecular targets that interconnect insulin and leptin signaling networks. In this regard, our study highlights the role of Rho GTPase protein Cdc42 as an important component of this complex network. Dysregulation of Cdc42, whether induced by external or internal factors, appears to be an important contributor to the development of leptin and insulin resistance. This dysregulation is prominent in a variety of metabolically active organs such as the brain, pancreas, adipose tissue, liver, and kidneys. The effects of Cdc42 have been confirmed in pancreatic β-cells and the hypothalamus, revealing striking expression patterns in glucose tolerance, insulin secretion, and insulin sensitivity. The association between increased Cdc42 activity and inhibition of autophagy in hepatocytes sheds light on potential mechanisms underlying obesity-related insulin resistance. In turn, Cdc42 may influence leptin secretion by mediating insulin action and Cdc42 dysfunction may be associated with leptin resistance. These discoveries open up promising therapeutic avenues for managing obesity and associated disorders. Potential interventions targeting Cdc42 activity hold promise for reducing obesity and enhancing leptin sensitivity. These novel approaches could revolutionize obesity treatments. However, owing to the tissue-specific nature of Cdc42 activity, it is imperative to tailor interventions aiming at different tissues and employ targeted delivery methods to minimize systemic side effects. Further in-depth studies are essential to thoroughly assess the effectiveness and safety of selective Cdc42 inhibitors.
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 23 January 2023).
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Abbott, J.H. Age, period and cohort effects on body mass index in New Zealand, 1997–2038. Aust. N. Z. J. Public Health 2018, 42, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Allman-Farinelli, M.A.; Chey, T.; Bauman, A.E.; Gill, T.; James, W.P.T. Age, period and birth cohort effects on prevalence of overweight and obesity in Australian adults from 1990 to 2000. Eur. J. Clin. Nutr. 2008, 62, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Diouf, I.; Charles, M.A.; Ducimetière, P.; Basdevant, A.; Eschwege, E.; Heude, B. Evolution of Obesity Prevalence in France: An Age-Period-Cohort Analysis. Epidemiology 2010, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Schramm, S.; Sørensen, T.I.A.; Davidsen, M.; Tolstrup, J.S. Changes in adult obesity prevalence in Denmark, 1987–2021: Age–period–cohort analysis of nationally representative data. Eur. J. Public Health 2023, 33, 463–467. [Google Scholar] [CrossRef] [PubMed]
- López-Gil, J.F.; Garcia-Hermoso, A.; Smith, L.; Firth, J.; Trott, M.; Mesas, A.E.; Jiménez-López, E.; Gutiérrez-Espinoza, H.; Tarraga-López, P.J.; Victoria-Montesinos, D. Global Proportion of Disordered Eating in Children and Adolescents A Systematic Review and Meta-analysis. JAMA Pediatr. 2023, 177, 363–372. [Google Scholar] [CrossRef]
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity May Accelerate the Aging Process. Front. Endocrinol. 2019, 10, 266. [Google Scholar] [CrossRef]
- Mizuno, T.; Shu, I.W.; Makimura, H.; Mobbs, C. Obesity Over the Life Course. Sci. Aging Knowl. Environ. 2004, 2004, re4. [Google Scholar] [CrossRef]
- Dobson, A.; Hockey, R.; Chan, H.-W.; Mishra, G. Flexible age-period-cohort modelling illustrated using obesity prevalence data. BMC Med. Res. Methodol. 2020, 20, 16. [Google Scholar] [CrossRef]
- Schwartz, S.M.; Kemnitz, J.W. Age- and gender-related changes in body size, adiposity, and endocrine and metabolic parameters in free-ranging rhesus macaques. Am. J. Phys. Anthropol. 1992, 89, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Kemnitz, J.W.; Howard, C.F., Jr. Obesity in free-ranging rhesus macaques. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1993, 17, 1–9. [Google Scholar]
- Robertson, I.D. The association of exercise, diet and other factors with owner-perceived obesity in privately owned dogs from metropolitan Perth, WA. Prev. Vet. Med. 2003, 58, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, J.M.; Donoghue, S.; Saidla, J.; Wills, J. Overweight cats: Prevalence and risk factors. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1994, 18 (Suppl. S1), S22–S28. [Google Scholar]
- Li, H.; Matheny, M.; Nicolson, M.; Türner, N.; Scarpace, P.J. Leptin gene expression increases with age independent of increasing adiposity in rats. Diabetes 1997, 46, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Bergen, H.; Kleopoulos, S.; Bauman, W.A.; Mobbs, C.V. Effects of nutritional status and aging on leptin gene expression in mice: Importance of glucose. Horm. Metab. Res. 1996, 28, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B.; Månsson, S.; Gingerich, R.L.; Havel, P.J. Regulation of plasma leptin in mice: Influence of age, high-fat diet, and fasting. Am. J. Physiol. 1997, 273, R113–R120. [Google Scholar] [CrossRef]
- Jacobson, L. Middle-aged C57BL/6 mice have impaired responses to leptin that are not improved by calorie restriction. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E786–E793. [Google Scholar] [CrossRef]
- Rusli, F.; Deelen, J.; Andriyani, E.; Boekschoten, M.V.; Lute, C.; van den Akker, E.B.; Müller, M.; Beekman, M.; Steegenga, W.T. Fibroblast growth factor 21 reflects liver fat accumulation and dysregulation of signalling pathways in the liver of C57BL/6J mice. Sci. Rep. 2016, 6, 30484. [Google Scholar] [CrossRef]
- Bazhan, N.M.; Baklanov, A.V.; Piskunova, J.V.; Kazantseva, A.J.; Makarova, E.N. Expression of genes involved in carbohydrate-lipid metabolism in muscle and fat tissues in the initial stage of adult-age obesity in fed and fasted mice. Physiol. Rep. 2017, 5, e13445. [Google Scholar] [CrossRef]
- Mobbs, C.V.; Moreno, C.L.; Poplawski, M. Metabolic mystery: Aging, obesity, diabetes, and the ventromedial hypothalamus. Trends Endocrinol. Metab. TEM 2013, 24, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, J.M.; Ros, M.; Andrés, A.; Fernández-Agulló, T.; Arribas, C. Changes in the neuroendocrine control of energy homeostasis by adiposity signals during aging. Exp. Gerontol. 2009, 44, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [PubMed]
- Pétervári, E.; Rostás, I.; Soós, S.; Tenk, J.; Mikó, A.; Füredi, N.; Székely, M.; Balaskó, M. Age versus nutritional state in the development of central leptin resistance. Peptides 2014, 56, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Muzumdar, R.; Yang, X.M.; Gabriely, I.; Berger, R.; Barzilai, N. Aging Is Associated with Resistance to Effects of Leptin on Fat Distribution and Insulin Action. J. Gerontol. Ser. A 2002, 57, B225–B231. [Google Scholar] [CrossRef]
- Carter, S.; Caron, A.; Richard, D.; Picard, F. Role of leptin resistance in the development of obesity in older patients. Clin. Interv. Aging 2013, 8, 829–844. [Google Scholar] [PubMed]
- Balaskó, M.; Soós, S.; Székely, M.; Pétervári, E. Leptin and aging: Review and questions with particular emphasis on its role in the central regulation of energy balance. J. Chem. Neuroanat. 2014, 61–62, 248–255. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and Opportunities of Defining Clinical Leptin Resistance. Cell Metab. 2012, 15, 150–156. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Matheny, M.; Moore, R.L.; Tümer, N. Impaired leptin responsiveness in aged rats. Diabetes 2000, 49, 431–435. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Matheny, M.; Shek, E.W. Impaired leptin signal transduction with age-related obesity. Neuropharmacology 2000, 39, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef]
- Lustig, R.H.; Collier, D.; Kassotis, C.; Roepke, T.A.; Kim, M.J.; Blanc, E.; Barouki, R.; Bansal, A.; Cave, M.C.; Chatterjee, S.; et al. Obesity I: Overview and molecular and biochemical mechanisms. Biochem. Pharmacol. 2022, 199, 115012. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.; Asakawa, A.; Amitani, H.; Inui, A. Stimulation of leptin secretion by insulin. Indian J. Endocrinol. Metab. 2012, 16, S543–S548. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Kolaczynski, J.W.; Polansky, M. Effects of prolonged hyperinsulinemia on serum leptin in normal human subjects. J. Clin. Investig. 1997, 100, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Doucet, E.; St-Pierre, S.; Alméras, N.; Mauriége, P.; Després, J.-P.; Richard, D.; Bouchard, C.; Tremblay, A. Fasting Insulin Levels Influence Plasma Leptin Levels Independently from the Contribution of Adiposity: Evidence from Both a Cross-Sectional and an Intervention Study1. J. Clin. Endocrinol. Metab. 2000, 85, 4231–4237. [Google Scholar] [CrossRef] [PubMed]
- Tsubai, T.; Noda, Y.; Ito, K.; Nakao, M.; Seino, Y.; Oiso, Y.; Hamada, Y. Insulin elevates leptin secretion and mRNA levels via cyclic AMP in 3T3-L1 adipocytes deprived of glucose. Heliyon 2016, 2, e00194. [Google Scholar] [CrossRef]
- Saad, M.F.; Khan, A.; Sharma, A.; Michael, R.; Riad-Gabriel, M.G.; Boyadjian, R.; Jinagouda, S.D.; Steil, G.M.; Kamdar, V. Physiological insulinemia acutely modulates plasma leptin. Diabetes 1998, 47, 544–549. [Google Scholar] [CrossRef]
- Kumar, R.; Mal, K.; Razaq, M.K.; Magsi, M.; Memon, M.K.; Memon, S.; Afroz, M.N.; Siddiqui, H.F.; Rizwan, A. Association of Leptin with Obesity and Insulin Resistance. Cureus 2020, 12, e12178. [Google Scholar] [CrossRef]
- Lustig, R.H.; Sen, S.; Soberman, J.E.; Velasquez-Mieyer, P.A. Obesity, leptin resistance, and the effects of insulin reduction. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2004, 28, 1344–1348. [Google Scholar] [CrossRef]
- Münzberg, H.; Myers, M.G., Jr. Molecular and anatomical determinants of central leptin resistance. Nat. Neurosci. 2005, 8, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Beg, M.; Kumar, D.; Shankar, K.; Varshney, S.; Rajan, S.; Srivastava, A.; Singh, K.; Sonkar, S.; Mahdi, A.A.; et al. Chronic hyper-leptinemia induces insulin signaling disruption in adipocytes: Implications of NOS2. Free Radic. Biol. Med. 2017, 112, 93–108. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.M.; Asadi, A.; Johnson, J.D.; Covey, S.D.; Kieffer, T.J. Leptin Deficiency in Rats Results in Hyperinsulinemia and Impaired Glucose Homeostasis. Endocrinology 2014, 155, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Saygılı, F.; Oge, A.; Yilmaz, C. Hyperinsulinemia and Insulin Insensitivity in Women with Nonclassical Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency: The Relationship between Serum Leptin Levels and Chronic Hyperinsulinemia. Horm. Res. 2005, 63, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, S.; Fanelli, C.; Paramore, D.; Brothers, J.; Landt, M. Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes 1996, 45, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamada, T.; Hosaka, S.; Kaneko, K.; Asai, Y.; Munakata, Y.; Seike, J.; Horiuchi, T.; Kodama, S.; Izumi, T.; et al. Inter-organ insulin-leptin signal crosstalk from the liver enhances survival during food shortages. Cell Rep. 2023, 42, 112415. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-Y.; Lai, X.-N.; Qian, X.-L.; Lv, L.-C.; Li, J.; Duan, J.; Xiao, X.-H.; Xiong, L.-X. Cdc42: A Novel Regulator of Insulin Secretion and Diabetes-Associated Diseases. Int. J. Mol. Sci. 2019, 20, 179. [Google Scholar] [CrossRef]
- Umbayev, B.; Safarova, Y.; Yermekova, A.; Nessipbekova, A.; Syzdykova, A.; Askarova, S. Role of a small GTPase Cdc42 in aging and age-related diseases. Biogerontology 2023, 24, 27–46. [Google Scholar] [CrossRef]
- Melendez, J.; Grogg, M.; Zheng, Y. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 2011, 286, 2375–2381. [Google Scholar] [CrossRef]
- Pichaud, F.; Walther, R.F.; Nunes de Almeida, F. Regulation of Cdc42 and its effectors in epithelial morphogenesis. J. Cell Sci. 2019, 132, jcs217869. [Google Scholar] [CrossRef]
- Fu, J.; Liu, B.; Zhang, H.; Fu, F.; Yang, X.; Fan, L.; Zheng, M.; Zhang, S. The role of cell division control protein 42 in tumor and non-tumor diseases: A systematic review. J. Cancer 2022, 13, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Molarius, A.; Lindén-Boström, M.; Granström, F.; Karlsson, J. Obesity continues to increase in the majority of the population in mid-Sweden—A 12-year follow-up. Eur. J. Public Health 2016, 26, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Shaper, A.G.; Mary, W. Overweight and obesity and weight change in middle aged men: Impact on cardiovascular disease and diabetes. J. Epidemiol. Community Health 2005, 59, 134. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Guo, H. Anorexia, undernutrition, weight loss, sarcopenia, and cachexia of aging. Eur. Rev. Aging Phys. Act. 2012, 9, 119–127. [Google Scholar] [CrossRef]
- Fakhouri, T.H.; Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of obesity among older adults in the United States, 2007–2010. NCHS Data Brief 2012, 106, 1–8. [Google Scholar]
- Keramat, S.A.; Alam, K.; Rana, R.H.; Chowdhury, R.; Farjana, F.; Hashmi, R.; Gow, J.; Biddle, S.J.H. Obesity and the risk of developing chronic diseases in middle-aged and older adults: Findings from an Australian longitudinal population survey, 2009–2017. PLoS ONE 2021, 16, e0260158. [Google Scholar] [CrossRef]
- Khan, S.S.; Krefman, A.E.; Zhao, L.; Liu, L.; Chorniy, A.; Daviglus, M.L.; Schiman, C.; Liu, K.; Shih, T.; Garside, D.; et al. Association of Body Mass Index in Midlife With Morbidity Burden in Older Adulthood and Longevity. JAMA Netw. Open 2022, 5, e222318. [Google Scholar] [CrossRef]
- Singh-Manoux, A.; Dugravot, A.; Shipley, M.; Brunner, E.J.; Elbaz, A.; Sabia, S.; Kivimaki, M. Obesity trajectories and risk of dementia: 28 years of follow-up in the Whitehall II Study. Alzheimer’s Dement. 2018, 14, 178–186. [Google Scholar] [CrossRef]
- Smith, S.R.; Lovejoy, J.C.; Greenway, F.; Ryan, D.; deJonge, L.; de la Bretonne, J.; Volafova, J.; Bray, G.A. Contributions of total body fat, abdominal subcutaneous adipose tissue compartments, and visceral adipose tissue to the metabolic complications of obesity. Metab. Clin. Exp. 2001, 50, 425–435. [Google Scholar] [CrossRef]
- Kralova Lesna, I.; Kralova, A.; Cejkova, S.; Fronek, J.; Petras, M.; Sekerkova, A.; Thieme, F.; Janousek, L.; Poledne, R. Characterisation and comparison of adipose tissue macrophages from human subcutaneous, visceral and perivascular adipose tissue. J. Transl. Med. 2016, 14, 208. [Google Scholar] [CrossRef]
- Suárez-Cuenca, J.A.; De La Peña-Sosa, G.; De La Vega-Moreno, K.; Banderas-Lares, D.Z.; Salamanca-García, M.; Martínez-Hernández, J.E.; Vera-Gómez, E.; Hernández-Patricio, A.; Zamora-Alemán, C.R.; Domínguez-Pérez, G.A.; et al. Enlarged adipocytes from subcutaneous vs. visceral adipose tissue differentially contribute to metabolic dysfunction and atherogenic risk of patients with obesity. Sci. Rep. 2021, 11, 1831. [Google Scholar] [CrossRef]
- Hunter, G.R.; Gower, B.A.; Kane, B.L. Age Related Shift in Visceral Fat. Int. J. Body Compos. Res. 2010, 8, 103–108. [Google Scholar] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- DiGirolamo, M.; Fine, J.B.; Tagra, K.; Rossmanith, R. Qualitative regional differences in adipose tissue growth and cellularity in male Wistar rats fed ad libitum. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1998, 274, R1460–R1467. [Google Scholar] [CrossRef] [PubMed]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- JafariNasabian, P.; Inglis, J.E.; Reilly, W.; Kelly, O.J.; Ilich, J.Z. Aging human body: Changes in bone, muscle and body fat with consequent changes in nutrient intake. J. Endocrinol. 2017, 234, R37–R51. [Google Scholar] [CrossRef]
- Shimokata, H.; Tobin, J.D.; Muller, D.C.; Elahi, D.; Coon, P.J.; Andres, R. Studies in the distribution of body fat: I. Effects of age, sex, and obesity. J. Gerontol. 1989, 44, M66–M73. [Google Scholar] [CrossRef]
- Hunter, G.; Lara-Castro, C.; Byrne, N.; Zakharkin, S.; Stonge, M.-P.; Allison, D. Weight Loss Needed to Maintain Visceral Adipose Tissue during Aging. Int. J. Body Compos. Res. 2005, 3, 55–61. [Google Scholar]
- Hunter, G.R.; Snyder, S.W.; Kekes-Szabo, T.; Nicholson, C.; Berland, L. Intra-abdominal adipose tissue values associated with risk of possessing elevated blood lipids and blood pressure. Obes. Res. 1994, 2, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Jensen, M.D. Metabolic changes in aging humans: Current evidence and therapeutic strategies. J. Clin. Investig. 2022, 132, e158451. [Google Scholar] [CrossRef] [PubMed]
- Newton, S.; Braithwaite, D.; Akinyemiju, T.F. Socio-economic status over the life course and obesity: Systematic review and meta-analysis. PLoS ONE 2017, 12, e0177151. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Yamada, Y.; Sagayama, H.; Ainslie, P.N.; Andersen, L.F.; Anderson, L.J.; Arab, L.; Baddou, I.; Bedu-Addo, K.; Blaak, E.E.; et al. Daily energy expenditure through the human life course. Science 2021, 373, 808–812. [Google Scholar] [CrossRef] [PubMed]
- van den Beld, A.W.; Kaufman, J.M.; Zillikens, M.C.; Lamberts, S.W.J.; Egan, J.M.; van der Lely, A.J. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. 2018, 6, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Cappola, A.R.; Auchus, R.J.; El-Hajj Fuleihan, G.; Handelsman, D.J.; Kalyani, R.R.; McClung, M.; Stuenkel, C.A.; Thorner, M.O.; Verbalis, J.G. Hormones and Aging: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2023, 108, 1835–1874. [Google Scholar] [CrossRef]
- Pataky, M.W.; Young, W.F.; Nair, K.S. Hormonal and Metabolic Changes of Aging and the Influence of Lifestyle Modifications. Mayo Clin. Proc. 2021, 96, 788–814. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Rydén, M. Human white adipose tissue: A highly dynamic metabolic organ. J. Intern. Med. 2022, 291, 611–621. [Google Scholar] [CrossRef]
- Veilleux, A.; Caron-Jobin, M.; Noël, S.; Laberge, P.Y.; Tchernof, A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes 2011, 60, 1504–1511. [Google Scholar] [CrossRef]
- Rydén, M.; Andersson, D.P.; Bergström, I.B.; Arner, P. Adipose tissue and metabolic alterations: Regional differences in fat cell size and number matter, but differently: A cross-sectional study. J. Clin. Endocrinol. Metab. 2014, 99, E1870–E1876. [Google Scholar] [CrossRef]
- Eriksson-Hogling, D.; Andersson, D.P.; Bäckdahl, J.; Hoffstedt, J.; Rössner, S.; Thorell, A.; Arner, E.; Arner, P.; Rydén, M. Adipose tissue morphology predicts improved insulin sensitivity following moderate or pronounced weight loss. Int. J. Obes. 2015, 39, 893–898. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and Insulin Receptors in Adipose Tissue Development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; McGraw, T.E.; Kahn, B.B. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. 2021, 33, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Claycombe, K.; Jones, B.; Standridge, M.; Guo, Y.S.; Chun, J.; Taylor, J.; Moustaid-Moussa, N. Insulin increases fatty acid synthase gene transcription in human adipocyte. Am. J. Physiol. 1998, 274, R1253–R1259. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.-L.; Manning, B.D. Insulin Stimulates Adipogenesis through the Akt-TSC2-mTORC1 Pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef]
- Geloen, A.; Collet, A.J.; Guay, G.; Bukowiecki, L.J. Insulin stimulates in vivo cell proliferation in white adipose tissue. Am. J. Physiol.-Cell Physiol. 1989, 256, C190–C196. [Google Scholar] [CrossRef]
- Klemm, D.J.; Leitner, J.W.; Watson, P.; Nesterova, A.; Reusch, J.E.; Goalstone, M.L.; Draznin, B. Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J. Biol. Chem. 2001, 276, 28430–28435. [Google Scholar] [CrossRef]
- Kuk, J.L.; Saunders, T.J.; Davidson, L.E.; Ross, R. Age-related changes in total and regional fat distribution. Ageing Res. Rev. 2009, 8, 339–348. [Google Scholar] [CrossRef]
- Haffner, S.M. Sex hormones, obesity, fat distribution, type 2 diabetes and insulin resistance: Epidemiological and clinical correlation. Int. J. Obes. 2000, 24, S56–S58. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Volkov, P.; Dayeh, T.; Esguerra, J.L.S.; Salö, S.; Eliasson, L.; Rönn, T.; Bacos, K.; Ling, C. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 2014, 15, 522. [Google Scholar] [CrossRef] [PubMed]
- Ciarambino, T.; Crispino, P.; Guarisco, G.; Giordano, M. Gender Differences in Insulin Resistance: New Knowledge and Perspectives. Curr. Issues Mol. Biol. 2023, 45, 7845–7861. [Google Scholar] [CrossRef]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Borissova, A.M.; Tankova, T.; Kirilov, G.; Koev, D. Gender-dependent effect of ageing on peripheral insulin action. Int. J. Clin. Pract. 2005, 59, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Karakelides, H.; Irving, B.A.; Short, K.R.; O’Brien, P.; Nair, K.S. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes 2010, 59, 89–97. [Google Scholar] [CrossRef]
- Traish, A.M.; Saad, F.; Guay, A. The dark side of testosterone deficiency: II. Type 2 diabetes and insulin resistance. J. Androl. 2009, 30, 23–32. [Google Scholar] [CrossRef]
- Barros, R.P.; Gustafsson, J. Estrogen receptors and the metabolic network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef]
- Carr, M.C. The Emergence of the Metabolic Syndrome with Menopause. J. Clin. Endocrinol. Metab. 2003, 88, 2404–2411. [Google Scholar] [CrossRef]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef]
- Erion, K.A.; Corkey, B.E. Hyperinsulinemia: A Cause of Obesity? Curr. Obes. Rep. 2017, 6, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Kurauti, M.A.; Soares, G.M.; Marmentini, C.; Bronczek, G.A.; Branco, R.C.S.; Boschero, A.C. Chapter Nine—Insulin and aging. In Vitamins and Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 115, pp. 185–219. [Google Scholar]
- Pyörälä, M.; Miettinen, H.; Laakso, M.; Pyörälä, K. Hyperinsulinemia and the Risk of Stroke in Healthy Middle-Aged Men. Stroke 1998, 29, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Kurauti, M.A.; Ferreira, S.M.; Soares, G.M.; Vettorazzi, J.F.; Carneiro, E.M.; Boschero, A.C.; Costa-Júnior, J.M. Hyperinsulinemia is associated with increasing insulin secretion but not with decreasing insulin clearance in an age-related metabolic dysfunction mice model. J. Cell. Physiol. 2019, 234, 9802–9809. [Google Scholar] [CrossRef] [PubMed]
- Møller, L.F.; Jespersen, J. Elevated insulin levels in men: An 11-year follow-up study. J. Cardiovasc. Risk 1995, 2, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.V.; Freeman, R.V.; Supiano, M.A.; Smith, M.J.; Galecki, A.T.; Halter, J.B. Glucose Metabolism in Older Adults: A Study Including Subjects More Than 80 Years of Age. J. Am. Geriatr. Soc. 1997, 45, 813–817. [Google Scholar] [CrossRef]
- Ribeiro, R.A.; Batista, T.M.; Coelho, F.M.; Boschero, A.C.; Lopes, G.S.; Carneiro, E.M. Decreased β-cell insulin secretory function in aged rats due to impaired Ca2+ handling. Exp. Physiol. 2012, 97, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.; Polonsky, K.S.; Beltz, W.F.; Wallace, P.; Brechtel, G.; Fink, R.I. Effects of Aging on Insulin Secretion. Diabetes 1989, 38, 1549–1556. [Google Scholar] [CrossRef]
- Marmentini, C.; Soares, G.M.; Bronczek, G.A.; Piovan, S.; Mareze-Costa, C.E.; Carneiro, E.M.; Boschero, A.C.; Kurauti, M.A. Aging Reduces Insulin Clearance in Mice. Front. Endocrinol. 2021, 12, 679492. [Google Scholar] [CrossRef]
- Fink, R.I.; Revers, R.R.; Kolterman, O.G.; Olefsky, J.M. The metabolic clearance of insulin and the feedback inhibition of insulin secretion are altered with aging. Diabetes 1985, 34, 275–280. [Google Scholar] [CrossRef]
- Pacini, G.; Beccaro, F.; Valerio, A.; Nosadini, R.; Crepaldi, G. Reduced beta-cell secretion and insulin hepatic extraction in healthy elderly subjects. J. Am. Geriatr. Soc. 1990, 38, 1283–1289. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, Obesity, and Inflammatory Age-Related Diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.; Singer, K. Obesity-induced inflammation: The impact of the hematopoietic stem cell niche. JCI Insight 2021, 6, e145295. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; Delproposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging Is Associated with an Increase in T Cells and Inflammatory Macrophages in Visceral Adipose Tissue. J. Immunol. 2011, 187, 6208–6216. [Google Scholar] [CrossRef] [PubMed]
- Ortega Martinez de Victoria, E.; Xu, X.; Koska, J.; Francisco, A.M.; Scalise, M.; Ferrante, A.W., Jr.; Krakoff, J. Macrophage content in subcutaneous adipose tissue: Associations with adiposity, age, inflammatory markers, and whole-body insulin action in healthy Pima Indians. Diabetes 2009, 58, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Rakotoarivelo, V.; Lacraz, G.; Mayhue, M.; Brown, C.; Rottembourg, D.; Fradette, J.; Ilangumaran, S.; Menendez, A.; Langlois, M.F.; Ramanathan, S. Inflammatory Cytokine Profiles in Visceral and Subcutaneous Adipose Tissues of Obese Patients Undergoing Bariatric Surgery Reveal Lack of Correlation with Obesity or Diabetes. EBioMedicine 2018, 30, 237–247. [Google Scholar] [CrossRef]
- Schmidt, F.M.; Weschenfelder, J.; Sander, C.; Minkwitz, J.; Thormann, J.; Chittka, T.; Mergl, R.; Kirkby, K.C.; Faßhauer, M.; Stumvoll, M.; et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PLoS ONE 2015, 10, e0121971. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Tesi, M.; Bugliani, M.; Ferri, G.; Suleiman, M.; De Luca, C.; Bosi, E.; Masini, M.; De Tata, V.; Gysemans, C.; Cardarelli, F.; et al. Pro-Inflammatory Cytokines Induce Insulin and Glucagon Double Positive Human Islet Cells That Are Resistant to Apoptosis. Biomolecules 2021, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Westerterp, M.; Murphy, A.J.; Subramanian, V.; Ferrante, A.W., Jr.; Tall, A.R.; Accili, D. Expanded granulocyte/monocyte compartment in myeloid-specific triple FoxO knockout increases oxidative stress and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Picó, C.; Palou, M.; Pomar, C.A.; Rodríguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Huising, M.O.; Kruiswijk, C.P.; Flik, G. Phylogeny and evolution of class-I helical cytokines. J. Endocrinol. 2006, 189, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Londraville, R.L.; Prokop, J.W.; Duff, R.J.; Liu, Q.; Tuttle, M. On the Molecular Evolution of Leptin, Leptin Receptor, and Endospanin. Front. Endocrinol. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Münzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metab. Clin. Exp. 2015, 64, 13–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef]
- Friedman, J.M. The Function of Leptin in Nutrition, Weight, and Physiology. Nutr. Rev. 2002, 60 (Suppl. S10), S1–S14. [Google Scholar] [CrossRef]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Löllmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 50–54. [Google Scholar] [CrossRef] [PubMed]
- Roszkowska-Gancarz, M.; Jonas, M.; Owczarz, M.; Kurylowicz, A.; Polosak, J.; Franek, E.; Slusarczyk, P.; Mossakowska, M.; Puzianowska-Kuznicka, M. Age-related changes of leptin and leptin receptor variants in healthy elderly and long-lived adults. Geriatr. Gerontol. Int. 2015, 15, 365–371. [Google Scholar] [CrossRef]
- Lavoie, O.; Michael, N.J.; Caron, A. A critical update on the leptin-melanocortin system. J. Neurochem. 2023, 165, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Morgan, P.J.; Trayhurn, P. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J. Neuroendocrinol. 1996, 8, 733–735. [Google Scholar] [CrossRef]
- Cheung, C.C.; Clifton, D.K.; Steiner, R.A. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 1997, 138, 4489–4492. [Google Scholar] [CrossRef]
- Münzberg, H.; Singh, P.; Heymsfield, S.B.; Yu, S.; Morrison, C.D. Recent advances in understanding the role of leptin in energy homeostasis. F1000Research 2020, 9, 451. [Google Scholar] [CrossRef]
- Huo, L.; Gamber, K.; Greeley, S.; Silva, J.; Huntoon, N.; Leng, X.H.; Bjørbaek, C. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 2009, 9, 537–547. [Google Scholar] [CrossRef]
- Biglari, N.; Gaziano, I.; Schumacher, J.; Radermacher, J.; Paeger, L.; Klemm, P.; Chen, W.; Corneliussen, S.; Wunderlich, C.M.; Sue, M.; et al. Functionally distinct POMC-expressing neuron subpopulations in hypothalamus revealed by intersectional targeting. Nat. Neurosci. 2021, 24, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.R.; Chuang, J.C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.; Ye, C.; Yang, Z.; Choi, B.; Chua, S., Jr.; Lowell, B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 2011, 71, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, P.G.F.; Wasinski, F.; Mansano, N.S.; Furigo, I.C.; Teixeira, P.D.S.; Gusmao, D.O.; Frazao, R.; Donato, J. Leptin Receptor Expression in GABAergic Cells is Not Sufficient to Normalize Metabolism and Reproduction in Mice. Endocrinology 2021, 162, bqab168. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; O’Brien, W.G., 3rd; Lee, C.C.; Myers, M.G., Jr.; Tong, Q. Role of GABA release from leptin receptor-expressing neurons in body weight regulation. Endocrinology 2012, 153, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Zeltser, L.M.; Seeley, R.J.; Tschöp, M.H. Synaptic plasticity in neuronal circuits regulating energy balance. Nat. Neurosci. 2012, 15, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 191–198. [Google Scholar] [CrossRef]
- Peng, J.; Yin, L.; Wang, X. Central and peripheral leptin resistance in obesity and improvements of exercise. Horm. Behav. 2021, 133, 105006. [Google Scholar] [CrossRef]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia Is Required for the Development of Leptin Resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef]
- Balland, E.; Cowley, M.A. New insights in leptin resistance mechanisms in mice. Front. Neuroendocrinol. 2015, 39, 59–65. [Google Scholar] [CrossRef]
- Filippi, B.M.; Lam, T.K. Leptin and aging. Aging 2014, 6, 82–83. [Google Scholar] [CrossRef]
- Isidori, A.M.; Strollo, F.; Morè, M.; Caprio, M.; Aversa, A.; Moretti, C.; Frajese, G.; Riondino, G.; Fabbri, A. Leptin and Aging: Correlation with Endocrine Changes in Male and Female Healthy Adult Populations of Different Body Weights. J. Clin. Endocrinol. Metab. 2000, 85, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-W.; Pan, W.-T.; Lee, Y.; Kakuma, T.; Zhou, Y.-T.; Unger, R.H. The role of leptin resistance in the lipid abnormalities of aging. FASEB J. 2001, 15, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Quirós Cognuck, S.; Reis, W.L.; Silva, M.; Debarba, L.K.; Mecawi, A.S.; de Paula, F.J.A.; Rodrigues Franci, C.; Elias, L.L.K.; Antunes-Rodrigues, J. Sex differences in body composition, metabolism-related hormones, and energy homeostasis during aging in Wistar rats. Physiol. Rep. 2020, 8, e14597. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Ogus, S.; Lu, R.; Chehab, F.F. Transgenic Mice Overexpressing Leptin Accumulate Adipose Mass at an Older, But Not Younger, Age*. Endocrinology 2001, 142, 348–358. [Google Scholar] [CrossRef]
- Schautz, B.; Later, W.; Heller, M.; Peters, A.; Müller, M.J.; Bosy-Westphal, A. Impact of age on leptin and adiponectin independent of adiposity. Br. J. Nutr. 2012, 108, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Belin de Chantemèle, E.J. Sex Differences in Leptin Control of Cardiovascular Function in Health and Metabolic Diseases. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Mauvais-Jarvis, F., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 87–111. [Google Scholar] [CrossRef]
- Shi, H. Gender difference in leptin production and leptin sensitivity. In Leptin: Hormonal Functions, Dysfunctions and Clinical Uses; Nova Science: Hauppauge, NY, USA, 2011; pp. 107–122. [Google Scholar]
- Guerra, B.; Fuentes, T.; Delgado-Guerra, S.; Guadalupe-Grau, A.; Olmedillas, H.; Santana, A.; Ponce-Gonzalez, J.G.; Dorado, C.; Calbet, J.A. Gender dimorphism in skeletal muscle leptin receptors, serum leptin and insulin sensitivity. PLoS ONE 2008, 3, e3466. [Google Scholar] [CrossRef] [PubMed]
- Van Harmelen, V.; Reynisdottir, S.; Eriksson, P.; Thörne, A.; Hoffstedt, J.; Lönnqvist, F.; Arner, P. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes 1998, 47, 913–917. [Google Scholar] [CrossRef]
- González-García, I.; García-Clavé, E.; Cebrian-Serrano, A.; Le Thuc, O.; Contreras, R.E.; Xu, Y.; Gruber, T.; Schriever, S.C.; Legutko, B.; Lintelmann, J.; et al. Estradiol regulates leptin sensitivity to control feeding via hypothalamic Cited1. Cell Metab. 2023, 35, 438–455.e7. [Google Scholar] [CrossRef]
- Elbers, J.M.; Asscheman, H.; Seidell, J.C.; Frölich, M.; Meinders, A.E.; Gooren, L.J. Reversal of the sex difference in serum leptin levels upon cross-sex hormone administration in transsexuals. J. Clin. Endocrinol. Metab. 1997, 82, 3267–3270. [Google Scholar] [CrossRef]
- Frederiksen, L.; Højlund, K.; Hougaard, D.M.; Mosbech, T.H.; Larsen, R.; Flyvbjerg, A.; Frystyk, J.; Brixen, K.; Andersen, M. Testosterone therapy decreases subcutaneous fat and adiponectin in aging men. Eur. J. Endocrinol. 2012, 166, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Behre, H.M.; Simoni, M.; Nieschlag, E. Strong association between serum levels of leptin and testosterone in men. Clin. Endocrinol. 1997, 47, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Maioli, S.; Leander, K.; Nilsson, P.; Nalvarte, I. Estrogen receptors and the aging brain. Essays Biochem. 2021, 65, 913–925. [Google Scholar] [CrossRef]
- Farhadi, Z.; Khaksari, M.; Azizian, H.; Dabiri, S. The brain neuropeptides and STAT3 mediate the inhibitory effect of 17-β Estradiol on central leptin resistance in young but not aged female high-fat diet mice. Metab. Brain Dis. 2022, 37, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Côté, I.; Green, S.M.; Toklu, H.Z.; Morgan, D.; Carter, C.S.; Tümer, N.; Scarpace, P.J. Differential physiological responses to central leptin overexpression in male and female rats. J. Neuroendocrinol. 2017, 29, e12552. [Google Scholar] [CrossRef]
- Rocha, M.; Bing, C.; Williams, G.; Puerta, M. Physiologic estradiol levels enhance hypothalamic expression of the long form of the leptin receptor in intact rats. J. Nutr. Biochem. 2004, 15, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.; Mitchell, T.D.; Hebert, S. Leptin-induced changes in body composition in high fat-fed mice. Exp. Biol. Med. 2003, 228, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Thomas, T.C.; Storlien, L.H.; Huang, X.F. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. 2000, 24, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.; Bowen, H.M.; Mitchell, T.D. Leptin resistance in mice is determined by gender and duration of exposure to high-fat diet. Physiol. Behav. 2003, 78, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Leptin and the Blood-Brain Barrier: Curiosities and Controversies. Compr. Physiol. 2021, 11, 2351–2369. [Google Scholar] [CrossRef]
- Magni, P.; Liuzzi, A.; Ruscica, M.; Dozio, E.; Ferrario, S.; Bussi, I.; Minocci, A.; Castagna, A.; Motta, M.; Savia, G. Free and bound plasma leptin in normal weight and obese men and women: Relationship with body composition, resting energy expenditure, insulin-sensitivity, lipid profile and macronutrient preference. Clin. Endocrinol. 2005, 62, 189–196. [Google Scholar] [CrossRef]
- Brabant, G.; Horn, R.; von zur Mühlen, A.; Mayr, B.; Wurster, U.; Heidenreich, F.; Schnabel, D.; Grüters-Kieslich, A.; Zimmermann-Belsing, T.; Feldt-Rasmussen, U. Free and protein bound leptin are distinct and independently controlled factors in energy regulation. Diabetologia 2000, 43, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Dallner, O.S.; Marinis, J.M.; Lu, Y.-H.; Birsoy, K.; Werner, E.; Fayzikhodjaeva, G.; Dill, B.D.; Molina, H.; Moscati, A.; Kutalik, Z.; et al. Dysregulation of a long noncoding RNA reduces leptin leading to a leptin-responsive form of obesity. Nat. Med. 2019, 25, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.K.; Opentanova, I.; Ohannesian, J.P.; Kolaczynski, J.W.; Heiman, M.L.; Hale, J.; Becker, G.W.; Bowsher, R.R.; Stephens, T.W.; Caro, J.F. Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short-term fasting. J. Clin. Investig. 1996, 98, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.E.; Panza, G.S.; Gollie, J.M. Leptin, Leptin Soluble Receptor, and the Free Leptin Index following a Diet and Physical Activity Lifestyle Intervention in Obese Males and Females. J. Obes. 2016, 2016, 8375828. [Google Scholar] [CrossRef] [PubMed]
- Maamra, M.; Bidlingmaier, M.; Postel-Vinay, M.C.; Wu, Z.; Strasburger, C.J.; Ross, R.J.M. Generation of Human Soluble Leptin Receptor by Proteolytic Cleavage of Membrane-Anchored Receptors. Endocrinology 2001, 142, 4389–4393. [Google Scholar] [CrossRef]
- Schaab, M.; Kratzsch, J. The soluble leptin receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 661–670. [Google Scholar] [CrossRef]
- Reinehr, T.; Kratzsch, J.; Kiess, W.; Andler, W. Circulating soluble leptin receptor, leptin, and insulin resistance before and after weight loss in obese children. Int. J. Obes. 2005, 29, 1230–1235. [Google Scholar] [CrossRef]
- Chan, J.L.; Blüher, S.; Yiannakouris, N.; Suchard, M.A.; Kratzsch, J.; Mantzoros, C.S. Regulation of Circulating Soluble Leptin Receptor Levels By Gender, Adiposity, Sex Steroids, and Leptin: Observational and Interventional Studies in Humans. Diabetes 2002, 51, 2105–2112. [Google Scholar] [CrossRef]
- van Dielen, F.M.; van ‘t Veer, C.; Buurman, W.A.; Greve, J.W. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J. Clin. Endocrinol. Metab. 2002, 87, 1708–1716. [Google Scholar] [CrossRef]
- Sun, Q.; van Dam, R.M.; Meigs, J.B.; Franco, O.H.; Mantzoros, C.S.; Hu, F.B. Leptin and Soluble Leptin Receptor Levels in Plasma and Risk of Type 2 Diabetes in U.S. Women: A Prospective Study. Diabetes 2009, 59, 611–618. [Google Scholar] [CrossRef]
- Drori, A.; Gammal, A.; Azar, S.; Hinden, L.; Hadar, R.; Wesley, D.; Nemirovski, A.; Szanda, G.; Salton, M.; Tirosh, B.; et al. CB1R regulates soluble leptin receptor levels via CHOP, contributing to hepatic leptin resistance. eLife 2020, 9, e60771. [Google Scholar] [CrossRef]
- Kratzsch, J.; Lammert, A.; Bottner, A.; Seidel, B.; Mueller, G.; Thiery, J.; Hebebrand, J.; Kiess, W. Circulating soluble leptin receptor and free leptin index during childhood, puberty, and adolescence. J. Clin. Endocrinol. Metab. 2002, 87, 4587–4594. [Google Scholar] [CrossRef] [PubMed]
- Quinton, N.D.; Smith, R.F.; Clayton, P.E.; Gill, M.S.; Shalet, S.; Justice, S.K.; Simon, S.A.; Walters, S.; Postel-Vinay, M.C.; Blakemore, A.I.F.; et al. Leptin Binding Activity Changes with Age: The Link between Leptin and Puberty1. J. Clin. Endocrinol. Metab. 1999, 84, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- Schaab, M.; Kausch, H.; Klammt, J.; Nowicki, M.; Anderegg, U.; Gebhardt, R.; Rose-John, S.; Scheller, J.; Thiery, J.; Kratzsch, J. Novel regulatory mechanisms for generation of the soluble leptin receptor: Implications for leptin action. PLoS ONE 2012, 7, e34787. [Google Scholar] [CrossRef] [PubMed]
- Uotani, S.; Bjørbaek, C.; Tornøe, J.; Flier, J.S. Functional properties of leptin receptor isoforms: Internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 1999, 48, 279–286. [Google Scholar] [CrossRef]
- Engeli, S. Dysregulation of the Endocannabinoid System in Obesity. J. Neuroendocrinol. 2008, 20, 110–115. [Google Scholar] [CrossRef]
- Matias, I.; Gonthier, M.-P.; Orlando, P.; Martiadis, V.; De Petrocellis, L.; Cervino, C.; Petrosino, S.; Hoareau, L.; Festy, F.; Pasquali, R.; et al. Regulation, Function, and Dysregulation of Endocannabinoids in Models of Adipose and β-Pancreatic Cells and in Obesity and Hyperglycemia. J. Clin. Endocrinol. Metab. 2006, 91, 3171–3180. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.P.; Peng, L.; Yuen, S.; Halstead, J.; Palacios, H.; Nyangau, E.; Mohammed, H.; Ziari, N.; Dandan, M.; Frakes, A.E.; et al. Aging alters the metabolic flux signature of the ER-unfolded protein response in vivo in mice. Aging Cell 2022, 21, e13558. [Google Scholar] [CrossRef]
- Morris, A. Mapping leptin-responsive neurons in the hypothalamus. Nat. Rev. Endocrinol. 2019, 15, 376–377. [Google Scholar] [CrossRef]
- Meister, B. Control of food intake via leptin receptors in the hypothalamus. Vitam. Horm. 2000, 59, 265–304. [Google Scholar] [CrossRef] [PubMed]
- Scarpace, P.J.; Matheny, M.; Tümer, N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience 2001, 104, 1111–1117. [Google Scholar] [CrossRef]
- Fernández-Galaz, C.; Fernández-Agulló, T.; Pérez, C.; Peralta, S.; Arribas, C.; Andrés, A.; Carrascosa, J.M.; Ros, M. Long-term food restriction prevents ageing-associated central leptin resistance in wistar rats. Diabetologia 2002, 45, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Pratap, A.A.; Holsinger, R.M.D. Altered Brain Leptin and Leptin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease. Pharmaceuticals 2020, 13, 401. [Google Scholar] [CrossRef] [PubMed]
- Guadalupe-Grau, A.; Larsen, S.; Guerra, B.; Calbet, J.A.L.; Dela, F.; Helge, J.W. Influence of age on leptin induced skeletal muscle signalling. Acta Physiol. 2014, 211, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Friedmann-Morvinski, D.; Alsaigh, T.; Kleifeld, O.; Kistler, E.B.; Rousso-Noori, L.; Huang, C.; Li, J.B.; Verma, I.M.; Schmid-Schönbein, G.W. Cleavage of the leptin receptor by matrix metalloproteinase–2 promotes leptin resistance and obesity in mice. Sci. Transl. Med. 2018, 10, eaah6324. [Google Scholar] [CrossRef] [PubMed]
- Cancemi, P.; Aiello, A.; Accardi, G.; Caldarella, R.; Candore, G.; Caruso, C.; Ciaccio, M.; Cristaldi, L.; Di Gaudio, F.; Siino, V.; et al. The Role of Matrix Metalloproteinases (MMP-2 and MMP-9) in Ageing and Longevity: Focus on Sicilian Long-Living Individuals (LLIs). Mediat. Inflamm. 2020, 2020, 8635158. [Google Scholar] [CrossRef]
- Chau, K.Y.; Sivaprasad, S.; Patel, N.; Donaldson, T.A.; Luthert, P.J.; Chong, N.V. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye 2007, 21, 1511–1515. [Google Scholar] [CrossRef]
- McNulty, M.; Spiers, P.; McGovern, E.; Feely, J. Aging is associated with increased matrix metalloproteinase-2 activity in the human aorta*. Am. J. Hypertens. 2005, 18, 504–509. [Google Scholar] [CrossRef]
- Boden, G.; Song, W.; Pashko, L.; Kresge, K. In vivo effects of insulin and free fatty acids on matrix metalloproteinases in rat aorta. Diabetes 2008, 57, 476–483. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Gu, B.; Cai, J.; Lv, Y.; Yu, L. Aerobic exercise regulates Rho/cofilin pathways to rescue synaptic loss in aged rats. PLoS ONE 2017, 12, e0171491. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Debidda, M.; Witte, D.; Zheng, Y. Cdc42 GTPase-activating protein deficiency promotes genomic instability and premature aging-like phenotypes. Proc. Natl. Acad. Sci. USA 2007, 104, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-R.; Kim, A.-R.; Kim, J.-S.; Kim, J.; Lee, J.-Y.; Lee, Y.-L.; Choe, M.; Park, J.-B. The proteins of synaptic vesicle membranes are affected during ageing of rat brain. Exp. Mol. Med. 2001, 33, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Afshordel, S.; Wood, W.G.; Igbavboa, U.; Muller, W.E.; Eckert, G.P. Impaired geranylgeranyltransferase-I regulation reduces membrane-associated Rho protein levels in aged mouse brain. J. Neurochem. 2014, 129, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Chaker, D.; Mouawad, C.; Azar, A.; Quilliot, D.; Achkar, I.; Fajloun, Z.; Makdissy, N. Inhibition of the RhoGTPase Cdc42 by ML141 enhances hepatocyte differentiation from human adipose-derived mesenchymal stem cells via the Wnt5a/PI3K/miR-122 pathway: Impact of the age of the donor. Stem Cell Res. Ther. 2018, 9, 167. [Google Scholar] [CrossRef]
- Umbayev, B.; Masoud, A.R.; Tsoy, A.; Alimbetov, D.; Olzhayev, F.; Shramko, A.; Kaiyrlykyzy, A.; Safarova, Y.; Davis, T.; Askarova, S. Elevated levels of the small GTPase Cdc42 induces senescence in male rat mesenchymal stem cells. Biogerontology 2018, 19, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Hwang, K.; Kim, Y.; Park, T. High-fat diet alters PP2A, TC10, and CIP4 expression in visceral adipose tissue of rats. Obesity 2008, 16, 1226–1231. [Google Scholar] [CrossRef]
- Miller, K.N.; Burhans, M.S.; Clark, J.P.; Howell, P.R.; Polewski, M.A.; DeMuth, T.M.; Eliceiri, K.W.; Lindstrom, M.J.; Ntambi, J.M.; Anderson, R.M. Aging and caloric restriction impact adipose tissue, adiponectin, and circulating lipids. Aging Cell 2017, 16, 497–507. [Google Scholar] [CrossRef]
- Kim, J.I.; Park, J.; Ji, Y.; Jo, K.; Han, S.M.; Sohn, J.H.; Shin, K.C.; Han, J.S.; Jeon, Y.G.; Nahmgoong, H.; et al. During Adipocyte Remodeling, Lipid Droplet Configurations Regulate Insulin Sensitivity through F-Actin and G-Actin Reorganization. Mol. Cell. Biol. 2019, 39, e00210-19. [Google Scholar] [CrossRef]
- Hansson, B.; Morén, B.; Fryklund, C.; Vliex, L.; Wasserstrom, S.; Albinsson, S.; Berger, K.; Stenkula, K.G. Adipose cell size changes are associated with a drastic actin remodeling. Sci. Rep. 2019, 9, 12941. [Google Scholar] [CrossRef]
- Rastogi, D.; Nico, J.; Johnston, A.D.; Tobias, T.A.M.; Jorge, Y.; Macian, F.; Greally, J.M. CDC42-related genes are upregulated in helper T cells from obese asthmatic children. J. Allergy Clin. Immunol. 2018, 141, 539–548.e537. [Google Scholar] [CrossRef]
- Keller, M.P.; Choi, Y.; Wang, P.; Davis, D.B.; Rabaglia, M.E.; Oler, A.T.; Stapleton, D.S.; Argmann, C.; Schueler, K.L.; Edwards, S.; et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008, 18, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamada, A.; Akimoto, Y.; Abe, K.; Matsubara, S.; Hayakawa, J.; Tanaka, J.; Kinoshita, M.; Kato, T.; Ogata, H.; et al. Cdc42 has important roles in postnatal angiogenesis and vasculature formation. Dev. Biol. 2021, 477, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Torisawa, Y.S.; Muyleart, M.; Hendee, K.; Anugwom, C.; Gutterman, D.; Mammoto, A. Effects of age-dependent changes in cell size on endothelial cell proliferation and senescence through YAP1. Aging 2019, 11, 7051–7069. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.K.; Yokoyama, M.; Yoshida, Y.; Nojima, A.; Kassai, H.; Oishi, K.; Okada, S.; Kinoshita, D.; Kobayashi, Y.; Fruttiger, M.; et al. A crucial role for CDC42 in senescence-associated inflammation and atherosclerosis. PLoS ONE 2014, 9, e102186. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.-J.; Shi, L.-L.; Liu, Y.; Wang, Y.; Bao, X.; Xu, W.; Yao, L.-Y.; Mbadhi, M.N.; Chen, L.; et al. Spatio-temporal model of Meox1 expression control involvement of Sca-1-positive stem cells in neointima formation through the synergistic effect of Rho/CDC42 and SDF-1α/CXCR4. Stem Cell Res. Ther. 2021, 12, 387. [Google Scholar] [CrossRef]
- Lengfeld, J.; Wang, Q.; Zohlman, A.; Salvarezza, S.; Morgan, S.; Ren, J.; Kato, K.; Rodriguez-Boulan, E.; Liu, B. Protein kinase C δ regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol. Biol. Cell 2012, 23, 1955–1963. [Google Scholar] [CrossRef]
- Xie, Y.; Potter, C.M.F.; Le Bras, A.; Nowak, W.N.; Gu, W.; Bhaloo, S.I.; Zhang, Z.; Hu, Y.; Zhang, L.; Xu, Q. Leptin Induces Sca-1+ Progenitor Cell Migration Enhancing Neointimal Lesions in Vessel-Injury Mouse Models. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2114–2127. [Google Scholar] [CrossRef]
- Yasmeen, M.F.; Cristobal, R.; Michele, S.; Jacqueline, M.; Yogi, P.; Puja, K.; John, V.; Bhama, R.; Chiara, B. Mapping the unicellular transcriptome of the ascending thoracic aorta to changes in mechanosensing and mechanoadaptation during aging. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ushakumary, M.G.; Wang, M.; Harikrishnan, V.; Titus, A.S.; Zhang, J.; Liu, L.; Monticone, R.; Wang, Y.; Mattison, J.A.; Cabo, R.; et al. Discoidin domain Receptor 2: A determinant of metabolic syndrome-associated arterial fibrosis in non-human primates. PLoS ONE 2019, 14, e0225911. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Zhang, X.; Ou, C.; Zhong, X.; Chen, Y.; Lu, L.; Liu, H.; Li, Y.; Liu, X.; et al. CDC42 promotes vascular calcification in chronic kidney disease. J. Pathol. 2019, 249, 461–471. [Google Scholar] [CrossRef]
- Ramchandran, R.; Mehta, D.; Vogel, S.M.; Mirza, M.K.; Kouklis, P.; Malik, A.B. Critical role of Cdc42 in mediating endothelial barrier protection in vivo. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 295, L363–L369. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Zhang, Y.; Tian, N.; Yang, J.; Ya, D.; Xiang, W.; Zhou, Z.; Jiang, Y.; Deng, J.; Yang, B.; et al. Leptin Promotes Angiogenesis via Pericyte STAT3 Pathway upon Intracerebral Hemorrhage. Cells 2022, 11, 2755. [Google Scholar] [CrossRef] [PubMed]
- Su, L.F.; Knoblauch, R.; Garabedian, M.J. Rho GTPases as modulators of the estrogen receptor transcriptional response. J. Biol. Chem. 2001, 276, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Zsakai, A.; Sipos, R.; Takacs-Vellai, K.; Szabo, A.; Bodzsar, E.B. The relationship between reproductive and biochemical ageing at the time of the menopausal transition. Exp. Gerontol. 2017, 98, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Azios, N.G.; Krishnamoorthy, L.; Harris, M.; Cubano, L.A.; Cammer, M.; Dharmawardhane, S.F. Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia 2007, 9, 147–158. [Google Scholar] [CrossRef]
- Papakonstanti, E.A.; Kampa, M.; Castanas, E.; Stournaras, C. A Rapid, Nongenomic, Signaling Pathway Regulates the Actin Reorganization Induced by Activation of Membrane Testosterone Receptors. Mol. Endocrinol. 2003, 17, 870–881. [Google Scholar] [CrossRef]
- Mimura, S.; Iwama, H.; Kato, K.; Nomura, K.; Kobayashi, M.; Yoneyama, H.; Miyoshi, H.; Tani, J.; Morishita, A.; Himoto, T.; et al. Profile of microRNAs associated with aging in rat liver. Int. J. Mol. Med. 2014, 34, 1065–1072. [Google Scholar] [CrossRef]
- Shchukina, I.; Bagaitkar, J.; Shpynov, O.; Loginicheva, E.; Porter, S.; Mogilenko, D.A.; Wolin, E.; Collins, P.; Demidov, G.; Artomov, M.; et al. Enhanced epigenetic profiling of classical human monocytes reveals a specific signature of healthy aging in the DNA methylome. Nat. Aging 2021, 1, 124–141. [Google Scholar] [CrossRef]
- Ding, L.; Zhang, L.; Kim, M.; Byzova, T.; Podrez, E. Akt3 kinase suppresses pinocytosis of low-density lipoprotein by macrophages via a novel WNK/SGK1/Cdc42 protein pathway. J. Biol. Chem. 2017, 292, 9283–9293. [Google Scholar] [CrossRef]
- Choi, S.-H.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein Accumulation in Macrophages via Toll-Like Receptor-4–Dependent Fluid Phase Uptake. Circ. Res. 2009, 104, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Jirholt, P.; Wramstedt, A.; Johansson, M.E.; Lundberg, A.M.; Trajkovska, M.G.; Ståhlman, M.; Fogelstrand, P.; Brisslert, M.; Fogelstrand, L.; et al. Rip2 Deficiency Leads to Increased Atherosclerosis Despite Decreased Inflammation. Circ. Res. 2011, 109, 1210–1218. [Google Scholar] [CrossRef]
- Florian, M.C.; Leins, H.; Gobs, M.; Han, Y.; Marka, G.; Soller, K.; Vollmer, A.; Sakk, V.; Nattamai, K.J.; Rayes, A.; et al. Inhibition of Cdc42 activity extends lifespan and decreases circulating inflammatory cytokines in aged female C57BL/6 mice. Aging Cell 2020, 19, e13208. [Google Scholar] [CrossRef] [PubMed]
- Møller, L.L.V.; Klip, A.; Sylow, L. Rho GTPases-Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells 2019, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Oh, E.; Thurmond, D.C. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem. 2007, 282, 9536–9546. [Google Scholar] [CrossRef] [PubMed]
- Yoder, S.M.; Dineen, S.L.; Wang, Z.; Thurmond, D.C. YES, a Src family kinase, is a proximal glucose-specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet β cells. J. Biol. Chem. 2014, 289, 11476–11487. [Google Scholar] [CrossRef] [PubMed]
- Usui, I.; Imamura, T.; Huang, J.; Satoh, H.; Olefsky, J.M. Cdc42 Is a Rho GTPase Family Member That Can Mediate Insulin Signaling to Glucose Transport in 3T3-L1 Adipocytes*. J. Biol. Chem. 2003, 278, 13765–13774. [Google Scholar] [CrossRef]
- Savova, M.S.; Mihaylova, L.V.; Tews, D.; Wabitsch, M.; Georgiev, M.I. Targeting PI3K/AKT signaling pathway in obesity. Biomed. Pharmacother. 2023, 159, 114244. [Google Scholar] [CrossRef]
- Veluthakal, R.; Chepurny, O.G.; Leech, C.A.; Schwede, F.; Holz, G.G.; Thurmond, D.C. Restoration of Glucose-Stimulated Cdc42-Pak1 Activation and Insulin Secretion by a Selective Epac Activator in Type 2 Diabetic Human Islets. Diabetes 2018, 67, 1999–2011. [Google Scholar] [CrossRef]
- Wang, Z.; Oh, E.; Clapp, D.W.; Chernoff, J.; Thurmond, D.C. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 2011, 286, 41359–41367. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, X.; Liu, Y.; Jiang, F.; Chen, M.; Cheng, L.; Cheng, X. Gene Expression Profiling of Type 2 Diabetes Mellitus by Bioinformatics Analysis. Comput. Math. Methods Med. 2020, 2020, 9602016. [Google Scholar] [CrossRef]
- Imamura, T.; Vollenweider, P.; Egawa, K.; Clodi, M.; Ishibashi, K.; Nakashima, N.; Ugi, S.; Adams, J.W.; Brown, J.H.; Olefsky, J.M. G alpha-q/11 protein plays a key role in insulin-induced glucose transport in 3T3-L1 adipocytes. Mol. Cell. Biol. 1999, 19, 6765–6774. [Google Scholar] [CrossRef] [PubMed]
- Cang, X.; Wang, Y.; Zeng, J.; Gao, J.; Yu, Q.; Lu, C.; Xu, F.; Lin, J.; Zhu, J.; Wang, X. C9orf72 knockdown alleviates hepatic insulin resistance by promoting lipophagy. Biochem. Biophys. Res. Commun. 2022, 588, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-H.; Li, J.-J. Aging and dyslipidemia: A review of potential mechanisms. Ageing Res. Rev. 2015, 19, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Bamba, V.; Rader, D.J. Obesity and atherogenic dyslipidemia. Gastroenterology 2007, 132, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, M.; Rough, S.J.; Alpert, J.S. Dyslipidemia in the elderly: Should it be treated? Clin. Cardiol. 2010, 33, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Lechner, K.; McKenzie, A.L.; Kränkel, N.; Von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingärtner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185. [Google Scholar] [CrossRef]
- Moro, E.; Gallina, P.; Pais, M.; Cazzolato, G.; Alessandrini, P.; Bittolo-Bon, G. Hypertriglyceridemia is associated with increased insulin resistance in subjects with normal glucose tolerance: Evaluation in a large cohort of subjects assessed with the 1999 World Health Organization criteria for the classification of diabetes. Metab. Clin. Exp. 2003, 52, 616–619. [Google Scholar] [CrossRef]
- Salonen, J.T.; Lakka, T.A.; Lakka, H.M.; Valkonen, V.P.; Everson, S.A.; Kaplan, G.A. Hyperinsulinemia is associated with the incidence of hypertension and dyslipidemia in middle-aged men. Diabetes 1998, 47, 270–275. [Google Scholar] [CrossRef]
- Mykkänen, L.; Kuusisto, J.; Haffner, S.M.; Pyörälä, K.; Laakso, M. Hyperinsulinemia predicts multiple atherogenic changes in lipoproteins in elderly subjects. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 518–526. [Google Scholar] [CrossRef]
- Steiner, G.; Vranic, M. Hyperinsulinemia and hypertriglyceridemia, a vicious cycle with atherogenic potential. Int. J. Obes. 1982, 6 (Suppl. S1), 117–124. [Google Scholar] [PubMed]
- Howard, B.V. Insulin resistance and lipid metabolism. Am. J. Cardiol. 1999, 84, 28j–32j. [Google Scholar] [CrossRef] [PubMed]
- Dannecker, C.; Wagner, R.; Peter, A.; Hummel, J.; Vosseler, A.; Häring, H.U.; Fritsche, A.; Birkenfeld, A.L.; Stefan, N.; Heni, M. Low-Density Lipoprotein Cholesterol Is Associated With Insulin Secretion. J. Clin. Endocrinol. Metab. 2021, 106, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Donald, C.; Jetha, A.; Covey, S.D.; Kieffer, T.J. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology 2010, 151, 4178–4186. [Google Scholar] [CrossRef] [PubMed]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metab. Clin. Exp. 2019, 93, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Fryk, E.; Olausson, J.; Mossberg, K.; Strindberg, L.; Schmelz, M.; Brogren, H.; Gan, L.-M.; Piazza, S.; Provenzani, A.; Becattini, B.; et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine 2021, 65, 103264. [Google Scholar] [CrossRef]
- Boden, G. Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2003, 111, 121–124. [Google Scholar] [CrossRef]
- Hawkins, M.; Barzilai, N.; Liu, R.; Hu, M.; Chen, W.; Rossetti, L. Role of the glucosamine pathway in fat-induced insulin resistance. J. Clin. Investig. 1997, 99, 2173–2182. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Plomgaard, P.; Akerstrom, T.; Møller, K.; Schmitz, O.; Pedersen, B.K. Effect of short-term intralipid infusion on the immune response during low-dose endotoxemia in humans. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E371–E379. [Google Scholar] [CrossRef]
- Einstein, F.H.; Huffman, D.M.; Fishman, S.; Jerschow, E.; Heo, H.J.; Atzmon, G.; Schechter, C.; Barzilai, N.; Muzumdar, R.H. Aging per se increases the susceptibility to free fatty acid-induced insulin resistance. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol. Metab. TEM 2010, 21, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free Fatty Acids Induce JNK-dependent Hepatocyte Lipoapoptosis*. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Sharma, M.; Urano, F.; Jaeschke, A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. J. Hepatol. 2012, 56, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef] [PubMed]
- Pretz, D.; Le Foll, C.; Rizwan, M.Z.; Lutz, T.A.; Tups, A. Hyperleptinemia as a contributing factor for the impairment of glucose intolerance in obesity. FASEB J. 2021, 35, e21216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e6. [Google Scholar] [CrossRef]
- Balland, E.; Chen, W.; Dodd, G.T.; Conductier, G.; Coppari, R.; Tiganis, T.; Cowley, M.A. Leptin Signaling in the Arcuate Nucleus Reduces Insulin’s Capacity to Suppress Hepatic Glucose Production in Obese Mice. Cell Rep. 2019, 26, 346–355.e3. [Google Scholar] [CrossRef]
- Kadowaki, T.; Hara, K.; Yamauchi, T.; Terauchi, Y.; Tobe, K.; Nagai, R. Molecular mechanism of insulin resistance and obesity. Exp. Biol. Med. 2003, 228, 1111–1117. [Google Scholar] [CrossRef]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. The role of leptin in the control of insulin-glucose axis. Front. Neurosci. 2013, 7, 51. [Google Scholar] [CrossRef]
- Matasha, D.; Gary, A.W.; Mingyan, Z.; Talley, J.L.; Monika, A.D.; Suzanne, M.A. Leptin-Induced Spine Formation Requires TrpC Channels and the CaM Kinase Cascade in the Hippocampus. J. Neurosci. 2014, 34, 10022. [Google Scholar] [CrossRef]
- Sahin, G.S.; Luis Rodriguez-Llamas, J.; Dillon, C.; Medina, I.; Appleyard, S.M.; Gaiarsa, J.L.; Wayman, G.A. Leptin increases GABAergic synaptogenesis through the Rho guanine exchange factor β-PIX in developing hippocampal neurons. Sci. Signal. 2021, 14, eabe4111. [Google Scholar] [CrossRef]
- Dhar, M.; Zhu, M.; Impey, S.; Lambert, T.J.; Bland, T.; Karatsoreos, I.N.; Nakazawa, T.; Appleyard, S.M.; Wayman, G.A. Leptin induces hippocampal synaptogenesis via CREB-regulated microRNA-132 suppression of p250GAP. Mol. Endocrinol. 2014, 28, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.P.; Tall, A.R. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J. Biol. Chem. 2001, 276, 49066–49076. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.; Hunter, J.C.; Javadov, S.; Karmazyn, M. mTOR mediates RhoA-dependent leptin-induced cardiomyocyte hypertrophy. Mol. Cell. Biochem. 2011, 352, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.; Hardy, S.C.; Irving, A.J.; Ashford, M.L. Leptin activation of ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cells involves disruption of the actin cytoskeleton. J. Physiol. 2000, 527 Pt 1, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Miller, L.C.; Laidlaw, H.A.; Burgess, L.A.; Perera, N.M.; Downes, C.P.; Leslie, N.R.; Ashford, M.L. A novel leptin signalling pathway via PTEN inhibition in hypothalamic cell lines and pancreatic beta-cells. EMBO J. 2006, 25, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G., Jr.; Schwartz, M.W.J.N. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef]
- Xu, A.W.; Kaelin, C.B.; Takeda, K.; Akira, S.; Schwartz, M.W.; Barsh, G.S. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J. Clin. Investig. 2005, 115, 951–958. [Google Scholar] [CrossRef]
- Barr, V.A.; Malide, D.; Zarnowski, M.J.; Taylor, S.I.; Cushman, S.W. Insulin Stimulates Both Leptin Secretion and Production by Rat White Adipose Tissue. Endocrinology 1997, 138, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Mastronardi, C.; Wong, M.L.; Licinio, J. Leptin therapy, insulin sensitivity, and glucose homeostasis. Indian J. Endocrinol. Metab. 2012, 16 (Suppl. S3), S549–S555. [Google Scholar] [CrossRef] [PubMed]
- Zeigerer, A.; Rodeheffer, M.S.; McGraw, T.E.; Friedman, J.M. Insulin regulates leptin secretion from 3T3-L1 adipocytes by a PI 3 kinase independent mechanism. Exp. Cell Res. 2008, 314, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.W.; Zhang, C.; Kahn, R.A.; Evans, T.; Cerione, R.A. Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J. Biol. Chem. 1996, 271, 26850–26854. [Google Scholar] [CrossRef] [PubMed]
- Serio, D.; Ispanovic, E.; Haas, T.L. Cdc42 increases activation of MMP-2 in endothelial cells. FASEB J. 2008, 22, 925.9. [Google Scholar] [CrossRef]
- Ispanovic, E.; Serio, D.; Haas, T. Cdc42 and RhoA have opposing roles in regulating membrane type 1-matrix metalloproteinase localization and matrix metalloproteinase-2 activation. Am. J. Physiol. Cell Physiol. 2008, 295, C600–C610. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective Hepatic Autophagy in Obesity Promotes ER Stress and Causes Insulin Resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Cumin, F.; Baum, H.P.; Levens, N. Leptin is cleared from the circulation primarily by the kidney. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1996, 20, 1120–1126. [Google Scholar]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome Analysis of Human Diabetic Kidney Disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef]
- Jiang, S.; Xu, C.-M.; Yao, S.; Zhang, R.; Li, X.-Z.; Zhang, R.-Z.; Xie, T.-Y.; Xing, Y.-Q.; Zhang, Q.; Zhou, X.-J.; et al. Cdc42 upregulation under high glucose induces podocyte apoptosis and impairs β-cell insulin secretion. Front. Endocrinol. 2022, 13, 905703. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Jensen, P.B.; Blum, W.F.; Christoffersen, C.T.; Englaro, P.; Heinze, E.; Rascher, W.; Teller, W.; Tornqvist, H.; Hauner, H. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes 1996, 45, 1435–1438. [Google Scholar] [CrossRef]
- Kolaczynski, J.W.; Nyce, M.R.; Considine, R.V.; Boden, G.; Nolan, J.J.; Henry, R.; Mudaliar, S.R.; Olefsky, J.; Caro, J.F. Acute and chronic effects of insulin on leptin production in humans: Studies in vivo and in vitro. Diabetes 1996, 45, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Fried, S.K. Multilevel regulation of leptin storage, turnover, and secretion by feeding and insulin in rat adipose tissue. J. Lipid Res. 2006, 47, 1984–1993. [Google Scholar] [CrossRef]
- Cong, L.; Chen, K.; Li, J.; Gao, P.; Li, Q.; Mi, S.; Wu, X.; Zhao, A.Z. Regulation of adiponectin and leptin secretion and expression by insulin through a PI3K-PDE3B dependent mechanism in rat primary adipocytes. Biochem. J. 2007, 403, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.S.; Wilken-Resman, B.; Williams, A.; DiLucia, S.; Sanchez, G.; McLeod, T.L.; Sims-Robinson, C. Hyperinsulinemia alters insulin receptor presentation and internalization in brain microvascular endothelial cells. Diabetes Vasc. Dis. Res. 2022, 19, 14791641221118626. [Google Scholar] [CrossRef]
- Fernández-Veledo, S.; Nieto-Vazquez, I.; de Castro, J.; Ramos, M.P.; Brüderlein, S.; Möller, P.; Lorenzo, M. Hyperinsulinemia Induces Insulin Resistance on Glucose and Lipid Metabolism in a Human Adipocytic Cell Line: Paracrine Interaction with Myocytes. J. Clin. Endocrinol. Metab. 2008, 93, 2866–2876. [Google Scholar] [CrossRef]
- Pu, Q.; Chang, Y.; Zhang, C.; Cai, Y.; Hassid, A. Chronic insulin treatment suppresses PTP1B function, induces increased PDGF signaling, and amplifies neointima formation in the balloon-injured rat artery. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H132–H139. [Google Scholar] [CrossRef]
- Wu, J.; Tao, W.; Bu, D.; Zhao, Y.; Zhang, T.; Chong, D.; Xue, B.; Xing, Z.; Li, C. Egr-1 transcriptionally activates protein phosphatase PTP1B to facilitate hyperinsulinemia-induced insulin resistance in the liver in type 2 diabetes. FEBS Lett. 2019, 593, 3054–3063. [Google Scholar] [CrossRef]
- Choi, J.; Ko, J.; Racz, B.; Burette, A.; Lee, J.R.; Kim, S.; Na, M.; Lee, H.W.; Kim, K.; Weinberg, R.J.; et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J. Neurosci. 2005, 25, 869–879. [Google Scholar] [CrossRef]
- Pallett, A.L.; Morton, N.M.; Cawthorne, M.A.; Emilsson, V. Leptin inhibits insulin secretion and reduces insulin mRNA levels in rat isolated pancreatic islets. Biochem. Biophys. Res. Commun. 1997, 238, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Seufert, J.; Kieffer, T.J.; Leech, C.A.; Holz, G.G.; Moritz, W.; Ricordi, C.; Habener, J.F. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: Implications for the development of adipogenic diabetes mellitus. J. Clin. Endocrinol. Metab. 1999, 84, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Hurley, J.D.; Smith, D.M.; Ghatei, M.A.; Withers, D.J.; Gardiner, J.V.; Bailey, C.J.; Bloom, S.R. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Investig. 1997, 100, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.T.; Lewis, J.T.; Cheung, A.T.; Luk, C.T.; Tse, J.; Wang, J.; Bryer-Ash, M.; Kolls, J.K.; Kieffer, T.J. Leptin Increases Hepatic Insulin Sensitivity and Protein Tyrosine Phosphatase 1B Expression. Mol. Endocrinol. 2004, 18, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Wilsey, J.; Scarpace, P.J. Caloric restriction reverses the deficits in leptin receptor protein and leptin signaling capacity associated with diet-induced obesity: Role of leptin in the regulation of hypothalamic long-form leptin receptor expression. J. Endocrinol. 2004, 181, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Wilsey, J.; Zolotukhin, S.; Prima, V.; Shek, E.W.; Matheny, M.K.; Scarpace, P.J. Hypothalamic delivery of doxycycline-inducible leptin gene allows for reversible transgene expression and physiological responses. Gene Ther. 2002, 9, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.-S.; Lee, H.-G.; Seo, J.-H.; Chung, C.-S.; Guo, D.-D.; Kim, T.-G.; Choi, Y.-J.; Cho, C.-S. Leptin-induced matrix metalloproteinase-2 secretion is suppressed by trans-10,cis-12 conjugated linoleic acid. Biochem. Biophys. Res. Commun. 2007, 356, 955–960. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Yoder, S.M.; Wang, Z.; Thurmond, D.C. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic β cells. Biochem. Pharmacol. 2013, 85, 808–816. [Google Scholar] [CrossRef]
- Baruah, M.P.; Kalra, S.; Ranabir, S. Metformin; A character actor in the leptin story! Indian J. Endocrinol. Metab. 2012, 16 (Suppl. S3), S532–S533. [Google Scholar] [CrossRef]
- Kumar, A.; Al-Sammarraie, N.; DiPette, D.J.; Singh, U.S. Metformin impairs Rho GTPase signaling to induce apoptosis in neuroblastoma cells and inhibits growth of tumors in the xenograft mouse model of neuroblastoma. Oncotarget 2014, 5, 11709–11722. [Google Scholar] [CrossRef]
- Ghorbani, Z.; Hekmatdoost, A.; Mirmiran, P. Anti-hyperglycemic and insulin sensitizer effects of turmeric and its principle constituent curcumin. Int. J. Endocrinol. Metab. 2014, 12, e18081. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Lankarani, K.B.; Tabrizi, R.; Ghayour-Mobarhan, M.; Peymani, P.; Ferns, G.; Ghaderi, A.; Asemi, Z. The Effects of Curcumin on Weight Loss Among Patients with Metabolic Syndrome and Related Disorders: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2019, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Jiao, D.M.; Yao, Q.H.; Yan, J.; Song, J.; Chen, F.Y.; Lu, G.H.; Zhou, J.Y. Expression analysis of Cdc42 in lung cancer and modulation of its expression by curcumin in lung cancer cell lines. Int. J. Oncol. 2012, 40, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Mongioì, L.M.; La Vignera, S.; Cannarella, R.; Cimino, L.; Compagnone, M.; Condorelli, R.A.; Calogero, A.E. The Role of Resveratrol Administration in Human Obesity. Int. J. Mol. Sci. 2021, 22, 4362. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Chen, L.-H.; Varadharajan, T.; Tsai, M.-J.; Chia, Y.-C.; Yuan, T.-C.; Sung, P.-J.; Weng, C.-F. Resveratrol inhibits glucose-induced migration of vascular smooth muscle cells mediated by focal adhesion kinase. Mol. Nutr. Food Res. 2014, 58, 1389–1401. [Google Scholar] [CrossRef]
- Albracht-Schulte, K.; Kalupahana, N.S.; Ramalingam, L.; Wang, S.; Rahman, S.M.; Robert-McComb, J.; Moustaid-Moussa, N. Omega-3 fatty acids in obesity and metabolic syndrome: A mechanistic update. J. Nutr. Biochem. 2018, 58, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chacińska, M.; Zabielski, P.; Książek, M.; Szałaj, P.; Jarząbek, K.; Kojta, I.; Chabowski, A.; Błachnio-Zabielska, A.U. The Impact of OMEGA-3 Fatty Acids Supplementation on Insulin Resistance and Content of Adipocytokines and Biologically Active Lipids in Adipose Tissue of High-Fat Diet Fed Rats. Nutrients 2019, 11, 835. [Google Scholar] [CrossRef]
- Sinha, S.; Haque, M.; Lugova, H.; Kumar, S. The Effect of Omega-3 Fatty Acids on Insulin Resistance. Life 2023, 13, 1322. [Google Scholar] [CrossRef]
- Gray, B.; Steyn, F.; Davies, P.S.W.; Vitetta, L. Omega-3 fatty acids: A review of the effects on adiponectin and leptin and potential implications for obesity management. Eur. J. Clin. Nutr. 2013, 67, 1234–1242. [Google Scholar] [CrossRef]
- Schmidt, S.; Willers, J.; Riecker, S.; Möller, K.; Schuchardt, J.P.; Hahn, A. Effect of omega-3 polyunsaturated fatty acids on the cytoskeleton: An open-label intervention study. Lipids Health Dis. 2015, 14, 4. [Google Scholar] [CrossRef]
- Turk, H.F.; Monk, J.M.; Fan, Y.-Y.; Callaway, E.S.; Weeks, B.; Chapkin, R.S. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am. J. Physiol. Cell Physiol. 2013, 304, C905–C917. [Google Scholar] [CrossRef]