


1. Introduction
Eurytrematosis, also known as pancreaticosis, is caused by parasitic trematodes belonging to the genus Eurytrema. These trematodes infect the pancreatic ducts of cattle, sheep, and other ruminants. In addition to infecting these livestock, certain species of these parasites can also infect humans [1,2]. Worms that stimulate the pancreatic duct can cause chronic proliferative inflammation and progressive necrosis of the mucosal epithelium. This, in turn, leads to the thickening of the tube wall, narrowing of the lumen, and possible occlusion [3,4]. Currently, over ten species of genus Eurytrema have been reported in the world, including E. pancreaticum, E. coelomaticum, E. cladorchis, E. dajii, E. ovis, E. tonkinense, E. parvum, and E. fukienensis [5]. However, some species within the genus Eurytrema have limited written descriptions, lacking anatomical structure maps, explanations of their life history, as well as other relevant information, such as gene sequences. Thus, there is a controversy regarding the validity for most of the type species [6]. E. pancreaticum, E. coelomaticum, and E. cladorchis are generally regarded as valid species and are considered the most predominant species in ruminants in China. These three parasites can coexist in the same region, especially in southern China. This coexistence creates difficulties in accurately identifying the Eurytrema species in the same region [7].
The mitochondrial genome possesses a distinctive genetic sequence that is inherited exclusively from the maternal line and exhibits a straightforward structure [5]. Due to its highly conserved coding region, elevated copy number, and limited recombination rate, the mitochondrial genome serves as a frequently utilized molecular marker in the examination of biological system evolution [8]. Its wide array of applications includes animal phylogeny, species identification, and parasite diagnosis, among other fields [9,10]. Up to date (December 2023), the GenBank database currently contains information only on three confirmed species within the Eurytrema genus: E. pancreaticum, E. coelomaticum, and E. cladorchis. Among these three species, only E. pancreaticum has an available complete mitochondrial genome sequence. As for other Eurytrema species, such as E. coelomaticum, sequencing has been limited to a few nuclear RNAs, including the 18S RNA gene. To accurately identify E. coelomaticum and provide additional molecular resources for further studies on Eurytrema taxonomy, population genetics, and systematics, we employed the Illumina NovaSeq sequencing platform and utilized the whole-genome shotgun strategy to create a library of the mitochondrial genome of E. coelomaticum.
2. Materials and Methods
2.1. Identification of Eurytrema coelomaticum
Samples were collected from the pancreatic organs of goats admitted to the Veterinary Hospital of Foshan University. The samples were stored in 70% alcohol at 4 °C. An adult worm was randomly selected, and its size was measured using a Vernier caliper. It was then stained with carmine. Its morphological characteristics were observed under a microscope, leading to its identification as E. coelomaticum based on the recently published morphological structure [11]. Additionally, it was identified as E. coelomaticum through the utilization of the 18S RNA and cox1 gene, as described in our previous study [12].
2.2. DNA Extraction and Sequencing
Genomic DNA extraction was performed using a genomic extraction kit provided by Nanjing Novizan Company on approximately 30 mg of parasite tissue that was cut into pieces. The concentration and purity of DNA were measured using a Thermo Scientific NanoDrop 2000 (Thermo Fisher Scientific Inc, the US), while DNA integrity was assessed using agarose gel electrophoresis and an Agilent 2100 Bioanalyzer. DNA samples that passed quality checks were subjected to shearing and fragmentation using a Covairs machine. Additionally, the ends of the DNA fragments were repaired using exonuclease and polymerase functions. The 3′ end of the splice contained a single T base, while a single A base was introduced into the 3′ end, allowing for complementary pairing of A and T to connect DNA fragments and splices. The splice containing the tag was incubated with ligase to connect it to the DNA fragment. The DNA fragments with connectors were then selectively enriched, and the DNA library was subsequently amplified. The library size was determined using an Agilent 2100 Bioanalyzer, while the total concentration of the library was measured through fluorescence quantitation. A single-chain library served as a template for bridging the polymerase chain reaction (PCR) amplification, synthesis, and sequencing. To validate the next-generation sequencing (NGS) result, PCR was conducted on the extracted DNA to amplify the cox1 gene, followed by Sanger sequencing. All polymerase chain reactions (PCRs) were conducted using the Taq PCR Master Mix Kit (Sangon Biotech, Shanghai, China) and a thermal cycler (Biometra TAdvanced 96 SG, Jena, Germany). Two sets of primers were used: 1) COX1A-F (5′-AGGTTAGGAGAGACTGTCTG-3′) and COX1A-R (5′-ACAAGCTGGAGCCAACAATC-3′), and 2) COX1B-F (5′-GTGTCTCCAGGTTTGATTCC-3′) and COX1B-R (5′-CGAATATCACACCCTACCAAC-3′). The PCR protocol consisted of an initial denaturation step at 94 °C for 3 min, followed by 36 cycles at 94 °C for 30 s, 52 °C for 40 s, and 72 °C for 45 s. A final extension was performed at 72 °C for 10 min.
2.3. Genome Sequence Assembly and Analysis
A5—Miseq v20150522 [13] and SPAdesv3.9.0 [14] were utilized for the assembly of the NGS data, starting from the initial stage, and for the construction of the contig and scaffold sequences. Sequences were extracted by considering the sequencing depth of the stitched sequences. Subsequently, sequences with high sequencing depths were subjected to BLASTN analysis using the nt library on NCBI (BLAST v2.2.31+), enabling the identification of mitochondrial sequences from each stitching result. By referring to the reference sequence and utilizing mummers v3.1 software, collinearity analysis was conducted to ascertain the positional relationships between contigs and facilitate contig gap filling [15]. The final mitochondrial sequence was obtained through the utilization of pilon v1.18 [16] to correct the obtained results.
2.4. Genome Annotation
The complete mitochondrial genome sequence was uploaded to MITOS for initial annotation and prediction of the secondary structure of RNA genes [17]. Additionally, the annotation results were manually corrected using SnapGene (V6.0.2) software and validated through homology alignments with other trematode species.
2.5. Phylogenetic Analysis
Mitochondrial genomes play a crucial role in the systematic classification of species. The combination of multiple genes can enhance the stability and reliability of phylogenetic reconstruction. To compare and analyze the mitochondrial genome dataset of 59 trematode species , we utilized the Phylosuite [18]. To ensure the accuracy of names and annotations, we initially normalized the sequences using both Phylosuite and manual approaches. We aligned the protein-coding genes using the MAFFT program [19] in normal alignment mode. After removing ambiguously aligned regions using Gblocks 0.91b [20], we concatenated the alignment results of all protein-coding genes into a joint dataset in PhyloSuite. Subsequently, the resulting files were analyzed using ModelFinder [21] to identify the most suitable evolutionary models for each gene, facilitating the maximum likelihood (ML) phylogenetic analyses. The maximum likelihood phylogeny was inferred using IQ-TREE [22] with the Partition model [23] of nucleotide substitution, and ultrafast bootstraps [24] with 100,000 replicates were performed. Additionally, the phylograms were visualized and annotated using iTOL v5 [25].
3. Results
3.1. Mitogenome Structure and Nucleotide Composition
The raw read sequence obtained through high-quality sequencing was 22,801,366 bp, and the clean read sequence obtained after filtration was 18,207,918 bp. Through gap sequencing, gene splicing, and assembly, we obtained the complete mitochondrial genome sequence of E. coelomaticum, which was 15,831 bp in length (GenBank accession: ON297668). The proportions of A, T, C, and G were 18.8%, 43.7%, 11.9%, and 25.6%, respectively, and the A+T content in E. coelomaticum was 62.5% . The genome annotation results revealed that the genome consisted of 36 genes, including 12 protein-coding genes (PCGs), two rRNA genes, and 22 tRNA genes. Additionally, there were two noncoding regions (Figure 1). The mitochondrial genome of E. coelomaticum contained 24 gene spacers in the genome sequence, ranging in length from 1 to 371 bp. Additionally, there were nine overlapping genes with lengths ranging from 1 to 63 bp .
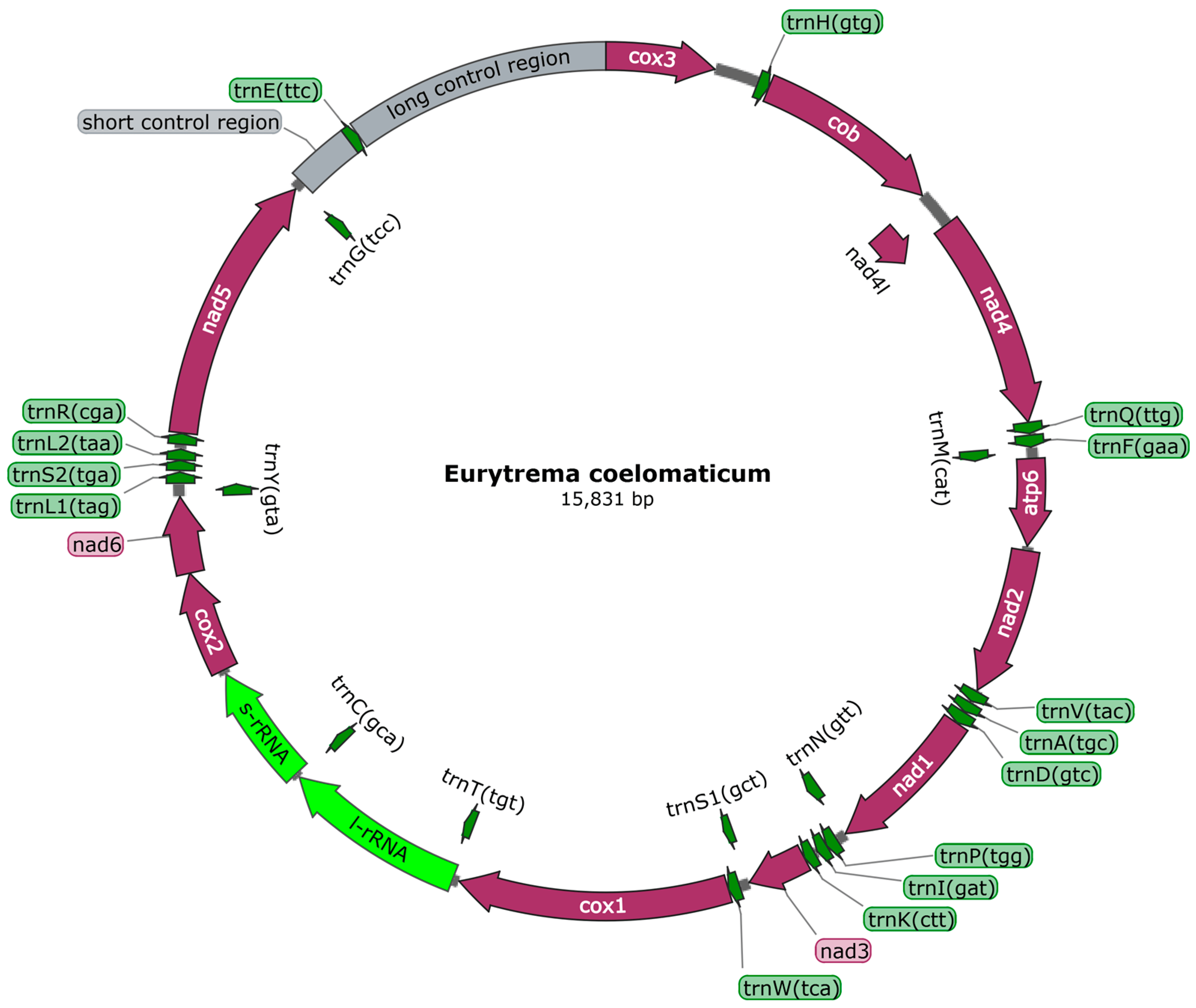
Figure 1. Eurytrema coelomaticum mitochondrial genome arrangement. All genes have a standard nomenclature. Twenty-two tRNA genes are designated by a letter code corresponding to amino acids and digitally distinguish between two specified leucine and serine tRNAs.
3.2. Protein-Coding Genes and Codon Usage
The 12 PCGs have a combined length of 10,302 bp, all oriented in the same translation direction and located on the sense sequence . Among the protein-coding genes, the cox1 gene is the longest, spanning 1579 bp (7183–8802), followed by the nad5 gene, which spans 1617 bp (12,159–13,775). The nad4L gene is the shortest, covering 264 bp (2107–2370). Nucleotide codon usage and codon family proportion are presented in and the top of the bar of Figure 2, and it is shown that the usage of T is very high and leucine (10.96% + 4.65% = 15.61%), Valine (11.92%), and phenylalanine (10.78%) were the most frequent amino acids in the PCGs of E. coelomaticum. The initiation codons of ten PCGs (nad5, nad6, cox1, nad3, nad1, nad2, atp6, nad4, nad4, and cob) are ATG, while the initiation codons of two PCGs (cox2 and cox3) are GTG. The stop codons of eight PCGs (atp6, cox2, cob, nad1, nad2, nad3, nad4l, and nad6) are TAA, the stop codons of four PCGs (cox1, cox3, nad4, and nad5) are TAG . To validate the NGS results and confirm the structure of the cox1 gene, two primers were designed for sequencing the start and end regions of the cox1 gene. It is shown that the cox1 gene starts at 7183 bp and ends at 8802 bp, encoding 539 aa (Figure 3). The length of the cox1 gene in the E. coelomaticum mitochondrial genome is the third longest among all currently sequenced family Dicrocoeliidae due to the presence of an additional amino acid tract at the N-terminal. A similar phenomenon is found in E. pancreaticum, which belongs to the same genus as E. coelomaticum .
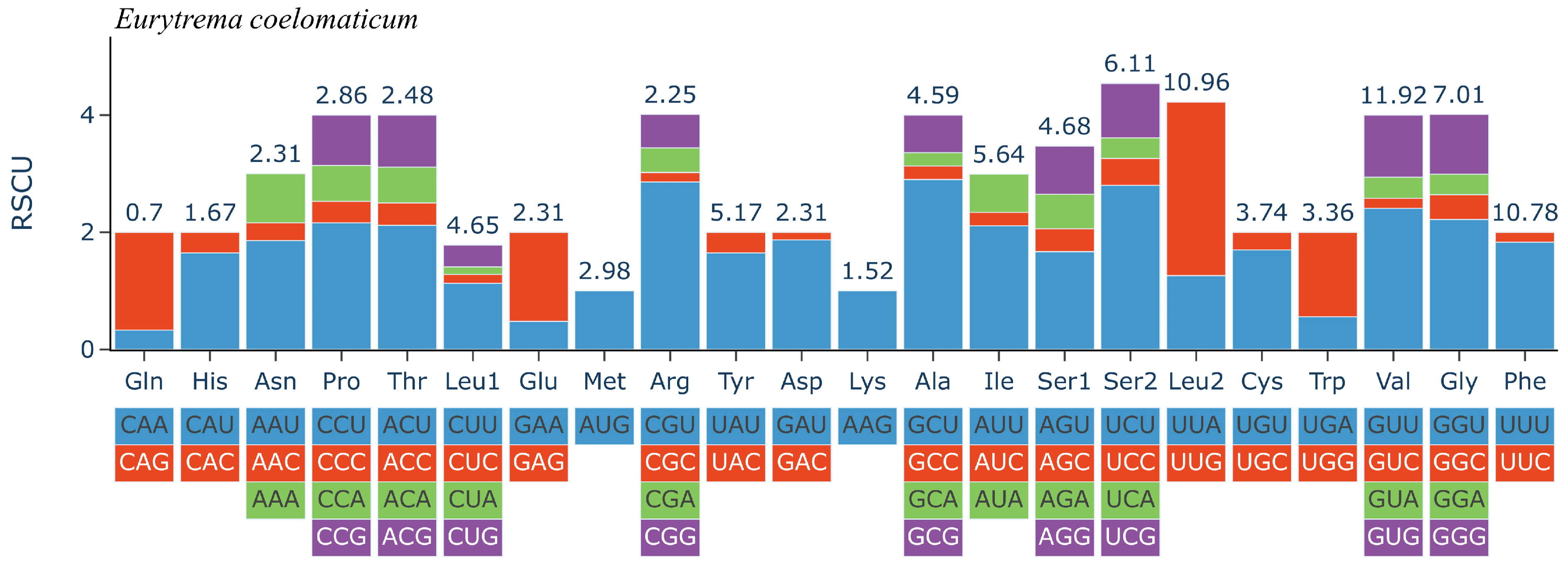
Figure 2. Relative synonymous codon usage (RSCU) of the complete mitochondrial genome of Eurytrema coelomaticum. Codon families are labelled on the x-axis. Values on the top of the bars refer to amino acid usage.
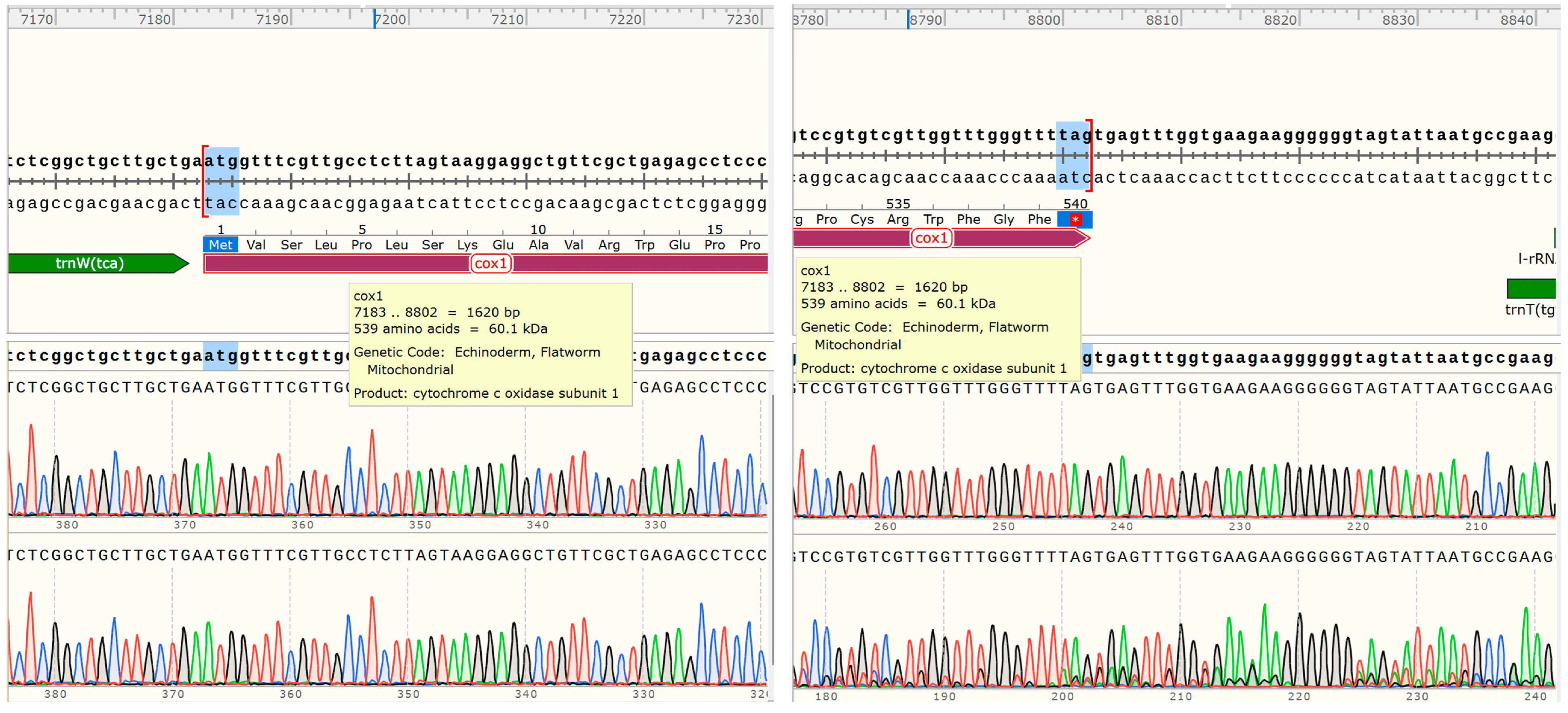
Figure 3. The cox1 gene location and length in E. coelomaticum mitochondrial genome.
3.3. tRNA and rRNA Genes
A total of 22 tRNAs, ranging in size from 60 to 80, were annotated in E. coelomaticum, with a total gene length of 1476 bp . The tRNA structures were predicted by MITOS, revealing that the structures of tRNA-S1 and tRNA-S2 were abnormal, presenting unorthodox structures in which the D-arms were unpaired . The remaining tRNAs exhibited the classical clover structure. The two rRNA genes are situated between the cox2 gene and the trnT gene, with a trnC gene separating them (Figure 1). The length of the rrnL gene is 1098 bp, while the rrnS gene is 722 bp in length. The A+T content of the rRNA gene sequence is 60.8% . Two noncoding regions (NCRs) with significant size differences were annotated in the mitochondrial genome of E. coelomaticum. The long noncoding region (LNCR) is 1564 bp in length and is situated between the trnE gene and the cox3 gene. The short noncoding region (SNCR) is 371 bp in length and is located between the trnG gene and the trnE gene. (Figure 1; . The A+T contents of NCRs are 66% and 69.7%, respectively .
3.4. Phylogenetic Analysis
The phylogenetic tree is constructed using the 12 PCGs from the concatenated regions of the mitochondrial genomes of 59 trematodes , with Gyrodactylus salaris (NC008815) serving as an outgroup. The maximum likelihood algorithm was used on the basis of the 9803 nucleic acid alignment length, available after Gblocks [20] processing. The phylogenetic tree analysis revealed that the 58 digeneans used in this study, including the suborders Diplostomata, Echinostomata, Haploporata, Hemiurata, Opisthorchiata, Pronocephalata, Troglotremata, and Xiphidiata, could be classified into two major clades. Clade I consists of three representative species of the family Schistosomatidae, while Clade II comprises 55 representatives from 28 families. Clade II includes E. coelomaticum and E. pancreaticum of the family Dicrocoeliidae, which are closely clustered and form a small clade (Figure 4). According to the phylogenetic tree, among the Digenea of Trematoda, the family Dicrocoeliidae is closely related to the families Prosthogonimidae, Eucotylidae, and clusters further with the families Plagiorchiidae, Orientocreadiidae, Haematoloechidae, and Glypthelminthidae within the suborder Xiphidiata. Additionally, based on the complete mitochondrial genome sequences of the used Digeneans, the genus Eurytrema is closely related to the genus Lyperosomum within the family Dicrocoeliidae (Figure 4). The entire family Dicrocoeliidae exhibits close clustering, despite significant evolutionary divergence.
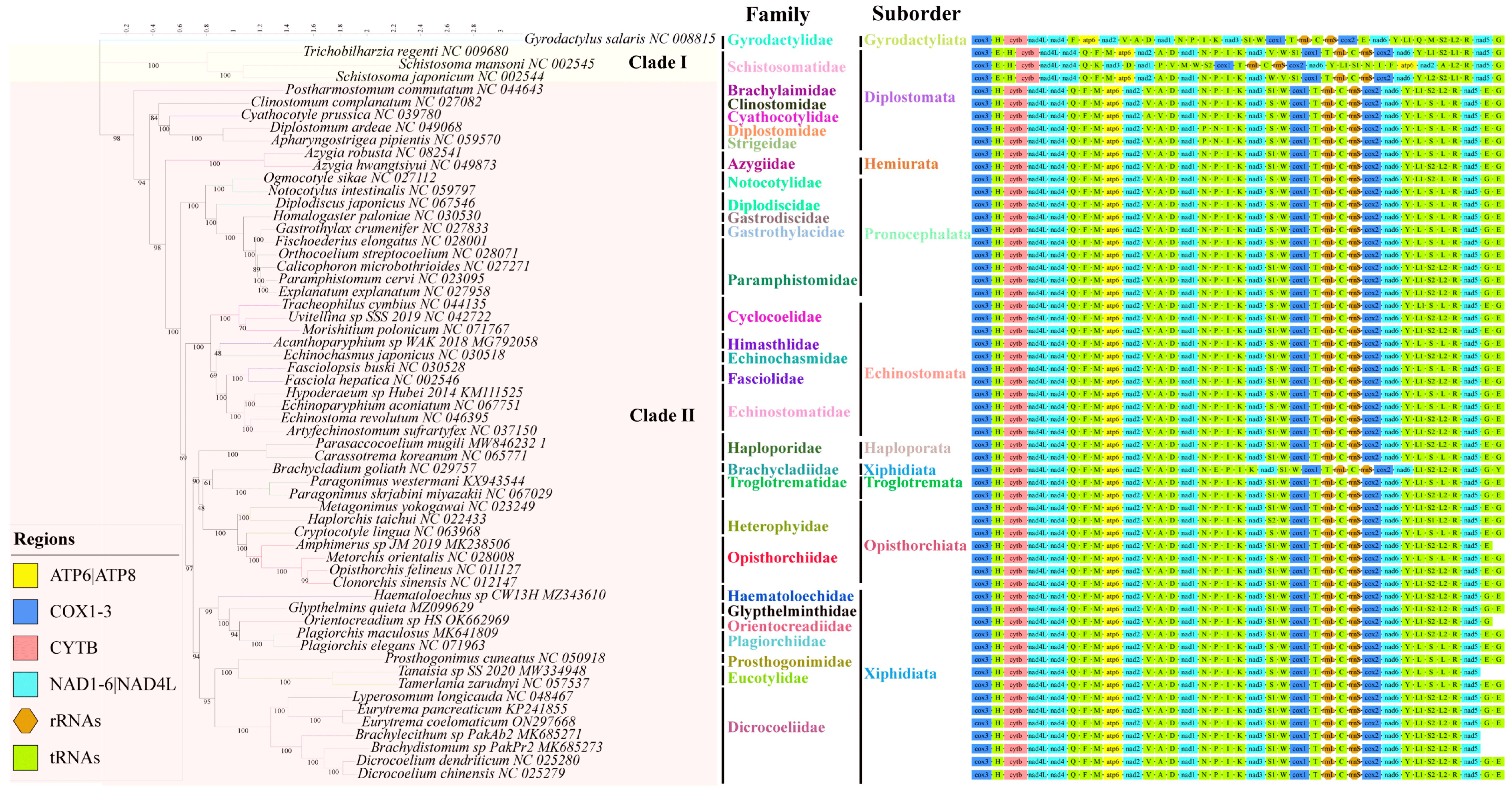
Figure 4. Phylogenetic relationships between Eurytrema coelomaticum and other trematodes based on mitochondrial sequences. Using the mitochondrial genome sequences of Gyrodactylus salaris (NC008815) as outgroup, the phylogenetic tree was constructed based on 12 protein-coding genes by the maximum likelihood (ML) method. The mitochondrial genome sequences of Brachylecithum sp. PakAb2, Brachydistomum sp. PakPr2, Tanaisia sp. SS-2020, Orientocreadium sp. HS, and Amphimerus sp. JM-2019 are incomplete.
4. Discussion
The species of the genus Eurytrema are common flukes in ruminants. However, the mitochondrial genome of some species within the genus Eurytrema are not well understood. Many gaps remain in the classification and evolutionary branching of Eurytrema. In this study, we sequenced and analyzed the complete mitochondrial genome sequence of E. coelomaticum.
The mitochondrial genome of E. coelomaticum has a total length of 15,831 bp, which is slightly longer than that of E. pancreaticum (15,031 bp) and most other flukes (e.g., 14,014 bp for Paramphistomum cervi (Accession: NC_023095.1) and 15,258 bp for Metagonimus yokogawai (Accession: NC_023249.1)). However, it falls within the range of mitochondrial genome lengths observed in sequenced trematodes. Notably, it is shorter than Schistosoma spindale (16,901 bp, Accession: NC_008067.1) and Tamerlania zarudnyi (16,188 bp, Accession: NC_057537.1). The complete mitochondrial genome of E. coelomaticum consists of 12 PCGs, two rRNAs, 22 tRNAs, and two noncoding regions. The initiation codons of PCGs in E. coelomaticum are similar to those found in E. pancreaticum and other Digenea trematodes, with the allocation of stop codons differing, using TAGs and TAAs [26,27]. Additionally, E. pancreaticum exhibits some PCGs with abbreviated T stop codons, whereas the sequenced mitochondrial genome of E. coelomaticum does not demonstrate this phenomenon. While it is common among metazoan mitochondrial protein genes, such abbreviated T stop codons have only been identified in cestodes among flatworms [26]. As for the cox1 gene, the maximum percentage variation in its protein size is 3.5% (512 aa–530 aa) in the mitochondrial DNA (mtDNA) of vertebrates and higher invertebrates such as insects and echinoderms [9]. The sequenced cox1 genes of trematodes in the RefSeq database show a range of sizes, from 510 aa (Fasciola hepatica, Accession: NP_066225) to 684 aa (Tamerlania zarudnyi, Accession: YP_010166578.1). Furthermore, within the genus Schistosoma, there is significant variation in the size of the cox1 gene, ranging from 513 aa to 609 aa [26,28,29]. In addition, the previous study shows that the structural abnormalities of tRNA-S1 and tRNA-S2 are normal in flatworms [30]. The long noncoding region (NCR) of E. pancreaticum is 989 bp in length [31], while that of E. coelomaticum measures 1564 bp. Trematodes of the same genus exhibit significant variations in NCR length, possibly due to the presence of a substantial number of repeats in the NCR of the mitochondria [32,33]. To address the issue of inaccurate NCR annotation, additional advancements in sequencing technology are required. The gene order of protein-coding and ribosomal RNA genes in the mitochondrial genome of E. coelomaticum conforms to the characteristic pattern observed in digeneans [30]. Furthermore, it was demonstrated that the positions of the last two genes, namely tRNA-Glu and tRNA-Gly, were interchanged across different species within Digenea, except for Schistosomatidae and the species of Brachycladium goliath (Figure 4).
In the past, studies on Eurytrema and even Dicrocoeliidae have mainly focused on morphology, life history, and prevalence, but little on the genetic evolution of genes. For decades, there has been considerable controversy over the taxonomy of Digenea. Previous studies on the phylogenetic analysis of Digenea and E. coelomaticum based on 28S rDNA and 18S rDNA sequences yielded different results [5,34,35]. Studies have shown that the mitochondrial genome sequence may provide useful genetic markers for examining the classification status of trematodes, especially when protein-coding gene sequences are used as comparative analysis markers [36,37]. In this study, phylogenetic analysis of mitochondrial genome sequences strongly suggests that the phylogeny of Digenea can be classified into two major clades. One clade consists of three species of Schistosomatidae, while the other consists of 55 members from 28 different families. Consistent with previous research [38], the evolutionary tree constructed in this study suggests that the suborders Xiphidiata and Diplostomata are paraphyletic, while other suborders are monophyletic and rooted in single branches. Additionally, in the Diplostomata branch, the families Clinostomidae, Cyathocotylidae, Dipostomidae, and Strigeidae cluster together, instead of being grouped with Schistosomatidae or Brachylaimidae (Figure 4). These results indicate that the Brachylaimidae, Clinostomidae, Cyathocotylidae, Dipostomidae, and Strigeidae taxonomies require further study. Within the suborder Xiphidiata, E. coelomaticum and E. pancreaticum are clustered together and are sister to Lyperosomum longicauda that form a small clade. This clade is more closely related to Dicrocoelium chinensis, D. dendriticum, Brachylecithum sp., and Brachydistomum sp. in the family Dicrocoeliidae than it is to species in Plagiorchiidae and Orientocreadiidae. Although Brachycladiidae is currently classified within the suborder Xiphidiata, an evolutionary tree constructed based on the species of Brachycladium goliath shows a closer relationship to Troglotrematidea, which is consistent with previous research, and indicated that Brachycladiidae and Troglotrematidae are more closely related to each other than either are to Dicrocoeliidae and Plagiorchiidae [38,39,40].
Our sequencing and annotation of the complete mitochondrial genome of E. coelomaticum makes a significant contribution to the Digenea database, specifically regarding the genus Eurytrema. Building upon these efforts, we reconstructed the phylogenetic tree of Digenea, validating the evolutionary relationships between E. coelomaticum and various Digenea flukes. This enhances our understanding of the overall phylogenetic relationship of Digenea.
5. Conclusions
This is the first reported complete mitochondrial genome of E. coelomaticum. The mitochondrial genome composition of E. coelomaticum is highly similar to that of E. pancreaticum, including 36 genes and two noncoding regions. Phylogenetic analysis showed that species of the genus Eurytrema and the genus Lyperosomum are more closely related to each other than to other trematodes. The mitochondrial data of E. coelomaticum can be a valuable resource for studying the evolutionary relationships within the genus Eurytrema and even within the family Dicrocoeliidae. Furthermore, it can provide a basis for the detection and systematic analysis of this parasite.
References
- Ishii, Y.; Koga, M.; Fujino, T.; Higo, H.; Ishibashi, J.; Oka, K.; Saito, S. Human infection with the pancreas fluke Eurytrema pancreaticum. Am. J. Trop. Med. Hyg. 1983, 32, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, D.E.R.; Barbosa, E.F.G.; Wilson, T.M.; Machado, M.; Oliveira, W.J.; Duarte, M.A.; Scalon, M.C.; Câmara, A.C.L.; Lux Hoppe, E.G.; Paludo, G.R.; et al. Eurytrema coelomaticum natural infection in small ruminants: A neglected condition. Parasitology 2021, 148, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Ilha, M.R.; Loretti, A.P.; Reis, A.C. Wasting and mortality in beef cattle parasitized by Eurytrema coelomaticum in the State of Paraná, southern Brazil. Vet. Parasitol. 2005, 133, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Rojo-Vázquez, F.A.; Meana, A.; Valcárcel, F.; Martínez-Valladares, M. Update on trematode infections in sheep. Vet. Parasitol. 2012, 189, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, U.K.; Ichikawa-Seki, M.; Hayashi, K.; Itagaki, T. Morphological and molecular characterization of Eurytrema cladorchis parasitizing cattle (Bos indicus) in Bangladesh. Parasitol. Res. 2015, 114, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Jones, A. Eurytrema cladorchis Chin, Li & Wei, 1965 (Trematoda: Dicrocoeliidae), a little known species from China and Nepal. Syst. Parasitol. 1985, 7, 43–45. [Google Scholar] [CrossRef]
- Song, M.; Zhang, L. Veterinary Parasitology; China Science Publishing Media Ltd.: Beijing, China, 2009. (In Chinese) [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Wang, P.; Yang, H.F.; Zhou, W.C.; Hwang, C.C.; Zhang, W.H.; Qian, Z.X. The mitochondrial genome of the land snail Camaena cicatricosa (Müller, 1774) (Stylommatophora, Camaenidae): The first complete sequence in the family Camaenidae. ZooKeys 2014, 451, 33–48. [Google Scholar] [CrossRef]
- Leite, K.G.; Lopes-Torres, E.J.; Souza, J.G.R.; Neves, R.H.; Gomes, D.C.; Machado-Silva, J.R. Eurytrema coelomaticum: Updated morphology of adult worms using advanced microscopy experiments. J. Helminthol. 2020, 94, e122. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, L.; Tian, S.; Zhang, B.; Liang, C.; Huang, F.; Liu, Q.; Zhang, H. Morphological Observation and Molecular Identification of Eurytrema coelmaticum in Goats. Progress. Vet. Med. 2019, 40, 34–38. (In Chinese) [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling Site Heterogeneity with Posterior Mean Site Frequency Profiles Accelerates Accurate Phylogenomic Estimation. Syst. Biol. 2018, 67, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Littlewood, D.T.; Lockyer, A.E.; Webster, B.L.; Johnston, D.A.; Le, T.H. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol. Phylogenet. Evol. 2006, 39, 452–467. [Google Scholar] [CrossRef]
- Liu, G.H.; Gasser, R.B.; Young, N.D.; Song, H.Q.; Ai, L.; Zhu, X.Q. Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasites Vectors 2014, 7, 150. [Google Scholar] [CrossRef]
- Jones, B.P.; Norman, B.F.; Borrett, H.E.; Attwood, S.W.; Mondal, M.M.H.; Walker, A.J.; Webster, J.P.; Rajapakse, R.; Lawton, S.P. Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale. Sci. Rep. 2020, 10, 2480. [Google Scholar] [CrossRef]
- Le, T.H.; Humair, P.F.; Blair, D.; Agatsuma, T.; Littlewood, D.T.; McManus, D.P. Mitochondrial gene content, arrangement and composition compared in African and Asian schistosomes. Mol. Biochem. Parasitol. 2001, 117, 61–71. [Google Scholar] [CrossRef]
- Le, T.H.; Blair, D.; McManus, D.P. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002, 18, 206–213. [Google Scholar] [CrossRef]
- Chang, Q.C.; Liu, G.H.; Gao, J.F.; Zheng, X.; Zhang, Y.; Duan, H.; Yue, D.M.; Fu, X.; Su, X.; Gao, Y.; et al. Sequencing and characterization of the complete mitochondrial genome from the pancreatic fluke Eurytrema pancreaticum (Trematoda: Dicrocoeliidae). Gene 2016, 576, 160–165. [Google Scholar] [CrossRef]
- Kinkar, L.; Young, N.D.; Sohn, W.M.; Stroehlein, A.J.; Korhonen, P.K.; Gasser, R.B. First record of a tandem-repeat region within the mitochondrial genome of Clonorchis sinensis using a long-read sequencing approach. PLoS Negl. Trop. Dis. 2020, 14, e0008552. [Google Scholar] [CrossRef] [PubMed]
- Kinkar, L.; Gasser, R.B.; Webster, B.L.; Rollinson, D.; Littlewood, D.T.J.; Chang, B.C.H.; Stroehlein, A.J.; Korhonen, P.K.; Young, N.D. Nanopore Sequencing Resolves Elusive Long Tandem-Repeat Regions in Mitochondrial Genomes. Int. J. Mol. Sci. 2021, 22, 1811. [Google Scholar] [CrossRef]
- Tkach, V.; Pawlowski, J.; Mariaux, J. Phylogenetic analysis of the suborder plagiorchiata (Platyhelminthes, Digenea) based on partial lsrDNA sequences. Int. J. Parasitol. 2000, 30, 83–93. [Google Scholar] [CrossRef]
- Figueira, G.F.; Oliveira, V.H.; Taroda, A.; Alfieri, A.A.; Headley, S.A. Molecular characterization of Eurytrema coelomaticum in cattle from Paraná, Brazil. Rev. Bras. De Parasitol. Vet. Braz. J. Vet. Parasitol. 2014, 23, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.B.; Wang, X.Y.; Lou, Z.Z.; Li, L.; Blair, D.; Yin, H.; Cai, J.Z.; Dai, X.L.; Lei, M.T.; Zhu, X.Q.; et al. The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. PLoS ONE 2013, 8, e71300. [Google Scholar] [CrossRef]
- Liu, G.H.; Yan, H.B.; Otranto, D.; Wang, X.Y.; Zhao, G.H.; Jia, W.Z.; Zhu, X.Q. Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol. 2014, 79, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.R.; Li, Y.; Gao, Y.; Qiu, Y.Y.; Jin, Z.H.; Gao, Z.Y.; Zhang, X.G.; An, Q.; Chang, Q.C.; Gao, J.F.; et al. The complete mitochondrial genome of Prosthogonimus cuneatus and Prosthogonimus pellucidus (Trematoda: Prosthogonimidae), their features and phylogenetic relationships in the superfamily Microphalloidea. Acta Trop. 2022, 232, 106469. [Google Scholar] [CrossRef]
- Briscoe, A.G.; Bray, R.A.; Brabec, J.; Littlewood, D.T. The mitochondrial genome and ribosomal operon of Brachycladium goliath (Digenea: Brachycladiidae) recovered from a stranded minke whale. Parasitol. Int. 2016, 65, 271–275. [Google Scholar] [CrossRef]
- Fu, Y.T.; Jin, Y.C.; Liu, G.H. The Complete Mitochondrial Genome of the Caecal Fluke of Poultry, Postharmostomum commutatum, as the First Representative from the Superfamily Brachylaimoidea. Front. Genet. 2019, 10, 1037. [Google Scholar] [CrossRef]
- Olson, P.D.; Cribb, T.H.; Tkach, V.V.; Bray, R.A.; Littlewood, D.T.J. Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int. J. Parasitol. 2003, 33, 733–755. [Google Scholar] [CrossRef]