


1. Introduction
The ever-growing emergence of antibiotic resistance associated with tuberculosis (TB) has become a global challenge [1]. Furthermore, the combination of TB and COVID-19 can cause severe respiratory complications, leading to systemic failure [2]. According to reports from the World Health Organization (WHO), TB was estimated to be the leading cause of death from a single infectious agent and the thirteenth leading cause of death worldwide [3]. However, in 2020 and 2021, it was projected that TB would rank second as a cause of death from a single infectious agent following the emergence of COVID-19 [4]. In 2021, it was estimated that approximately 10.6 million individuals worldwide contracted tuberculosis (TB); out of this total, it is estimated that 191,000 deaths occurred specifically due to multidrug-resistant or rifampicin-resistant tuberculosis (MDR/RR-TB) [5].
For the management of MDR-TB, WHO suggests two-course durations, which are abbreviated as short (9–11 months) and long (18–20 months) treatment regimens [2,6]. However, the concomitant use of multiple anti-TB medications and lengthy treatment regimens is often met with poor patient adherence, resulting in the emergence of Mycobacterium strains, which are resistant to first-line drugs such as isoniazid and rifampicin, leading to one of the most serious kinds of tuberculosis, i.e., MDR-TB. This has spurred the necessity of research and development of new drugs to fight against TB [7,8]. Albeit, various regulatory bodies are working to overcome the burden of this disease. The USFDA gave expedited approval to bedaquiline (TMC207) in 2012 as a new treatment for drug-resistant TB in adults when there are no other viable options available [9].
Bedaquiline (BDQ), a diarylquinoline derivative, has a unique mechanism of action. The drug specifically targets and deactivates the F1/F0-ATP synthase of MDR strains of Mycobacterium tuberculosis (M. tb) by binding to its subunit C of ATP synthase enzyme, which is responsible for energy production in M. tb without interfering with the F1/F0-ATP synthase in mammalian cells [10,11]. In preclinical and clinical studies, bedaquiline has shown promising results in treating MDR-TB. These studies involved the use of bedaquiline as a single drug or in combination with other anti-TB drugs (e.g., moxifloxacin and pyrazinamide). Additionally, some studies utilized a murine model of TB, and the findings support the efficacy of bedaquiline in treating MDR-TB [12]. In these studies, bedaquiline was administered at a dosage of 400 mg once daily for a period of 2 weeks, followed by 200 mg three times weekly for either 6 or 22 weeks, and showed improved treatment outcomes when compared with existing treatment regimens. Also, the bedaquiline regimen led to faster culture conversion (when the bacteria are no longer detectable in the patient’s sputum); this suggests that treatment duration with bedaquiline regimens may be shorter compared to other standard regimens [13].
Despite this unique anti-TB action of this drug, it is also associated with certain physiochemical limitations. According to the Biopharmaceutical Classification System (BCS), bedaquiline belongs to the Class II category, as it has exceptionally low solubility in water (0.000193 mg/mL) and high lipophilicity (log P 7.1) [14], thus making it an unsuitable molecule for conventional dosage forms. Furthermore, bedaquiline carries with it several dose-dependent potential adverse effects, such as prolonged QT interval, increased hepatic aminotransferase levels, nausea, vomiting, etc., which limit its use [15]. There are several formulation strategies, such as reduction in size, complexation, polymorphism, and solid dispersions, which overcome the hurdles of dose-dependent adverse effects, solubility, and release rate but do not resolve the bioavailability problems stemming from gastric degradation.
Of late, researchers have found self-nano-emulsifying drug delivery systems (SNEDDSs) to be the most advantageous lipid-based drug delivery systems for circumventing the existing challenges. They have also gained tremendous popularity due to their advantageous features, like small globule size, simplified production process, increased biocompatibility, and enhanced stability [16]. These systems are typically composed of lipids (natural and synthetic oils), surfactants, co-surfactants, and/or co-solvents that spontaneously emulsify. By pre-dissolving the drug in a mixture of lipids and emulsifying excipients, the disintegration and dissolution steps can be bypassed, which usually hinders the oral absorption of water-insoluble drugs [17].
The systematic use of design of experiments (DoE) to optimize isotropic systems has become common practice in both industry and academia [18]. The recent approach of “formulation by design” (FbD), based on DoE and quality by design (QbD), provides a rational understanding of the interaction between variables and helps to select the best formulation with minimal time, effort, and cost compared to the traditional one-factor-at-a-time (OFAT) approach [18]. The FbD methodology involves defining the quality target product profile (QTPP), identification of critical quality attributes (CQAs), critical material attributes (CMAs), and critical process parameters (CPPs) through screening and risk assessment, optimization data analysis using DoE, modeling and optimum search through response surface methodology (RSM) to create the design space, and developing a control strategy for continuous improvement [19]. The previous experimental studies carried out in various laboratories on diverse nanostructured systems, like liquid and S-SNEDDS of carvedilol [20] and ezetimibe [21], solid lipid nanoparticles of quercetin [22], isotretinoin [23], and liposomes of nimesulide [24], have vouched for the utility of QbD in developing the optimized nanocolloidal formulations.
The novelty of the current research work is related to the preparation of SNEDDS loaded with bedaquiline fumarate through the systematic optimization of the concentration of oil, surfactant, and co-surfactant. Furthermore, the study aims to investigate the biopharmaceutical performance of the developed SNEDDS, including in vitro release and stability studies, to confirm their retainability and quality under various physiological conditions. Finally, to assess the cytotoxicity of the optimized formulation, an MTT assay was conducted on A549 cells.
2. Results and Discussion
2.1. Solubility Study
The maximum solubility of BDQ-F in oils was found in caprylic acid (6.27 ± 10.90 mg/mL) and among the surfactants and co-surfactants, propylene glycol and Transcutol-P exhibited the highest solubility (6 ± 2.10 mg/mL), as shown in Figure 1 and Figure 2. Caprylic acid is a medium-chain saturated fatty acid, and the free fatty acid significantly increases the solubility of the drug by showcasing it as a lipophilic solubilizer [25]. Propylene glycol is known to reduce the interfacial tension of oil in water [26]. Transcutol-P has an HLB value of 4.2 and the ability to permeate GI mucosa by disrupting the lipid bilayer and enhancing oral bioavailability.
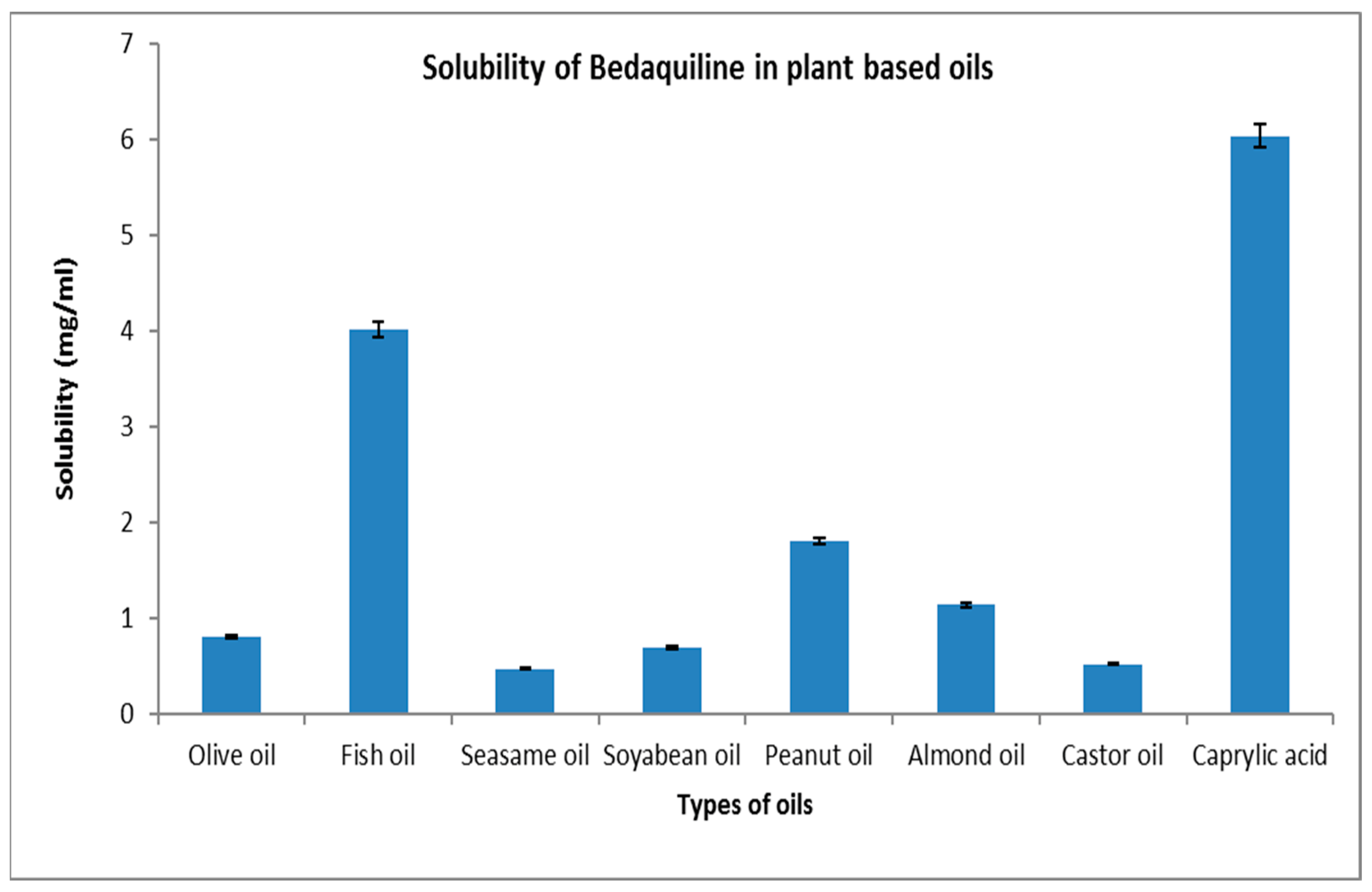
Figure 1. Solubility of BDQ-F in lipids.
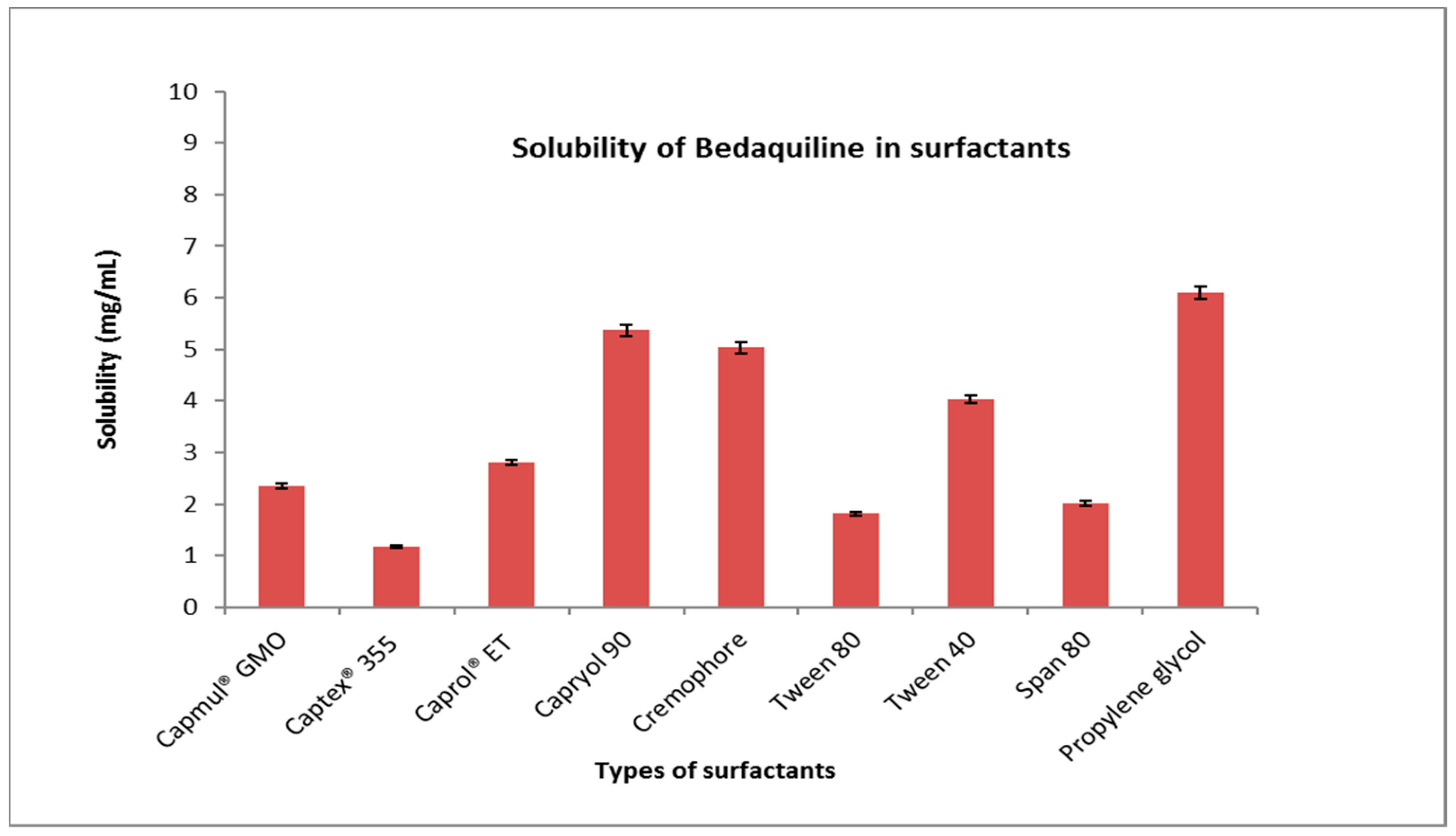
Figure 2. Solubility of BDQ-F in surfactants.
2.2. Pseudoternary Phase Diagrams
Pseudoternary phase diagrams were created to determine the optimal surfactant: co-surfactant (Smix) and oil: Smix ratios for SNEDDS development, as shown in . The pink dots represent the nanoemulsion region. It was observed that when a 1:0 Smix ratio was used, the nanoemulsion region obtained was almost negligible because propylene glycol was used alone, and sufficient emulsification of oil was not achieved. When the co-surfactant was incorporated in a Smix ratio of 1:1, the nanoemulsion region increased slightly. On increasing the quantity of co-surfactant in the Smix ratio 1:2 further, no increase in the nanoemulsion region was observed. On the other hand, when the concentration of surfactant quantity was increased from 1:1 to 2:1, an increase in the nanoemulsion region was observed, and 13.71% (v/v) oil was found to be emulsified with 41 % v/v of Smix. On further increasing the quantity of surfactant in the Smix 3:1 ratio, there was an appreciable increase in the nanoemulsion region, and the maximum amount of oil that could be emulsified was found to be 12.90 % v/v using 51.60 % v/v of Smix. For the Smix 4:1 and 5:1 ratios, there was no appreciable increase in the nanoemulsion region. Based on the pseudoternary diagram concentrations of oil, Smix was taken between the ranges of 10–30% and 40–60% [27].
2.3. Optimization Using BBD
The final optimization was conducted using Design Expert® software version 10.0.4 by Stat-Ease in Minneapolis, USA) using caprylic acid (A) (as oil), Smix (B) (consisting of propylene glycol, as a surfactant), Transcutol-P (as a co-surfactant), and the sonication time (C) as independent variables. The size of droplets (measured in nanometers), PDI, and percentage of transmittance were taken as dependent variables. Responses to fourteen runs, as shown in , were used to determine the significant model. The polynomial equations obtained for droplet size, PDI, and transmittance are as follows:
+ 0.5500AC + 1.00BC + 15.36A2 + 0.0563B2 + 6.14C2
− 0.0328BC + 0.0129A2 − 0.0101B2 + 0.0329C2
− 0.1000AC − 0.0500BC − 0.8625A2 − 0.0625B2 − 0
2.4. Validation and Point Prediction
On the basis of observed values of variables and the constraints applied, the software generated a predicted response for the optimal formulation, and this response was then compared with the experimental/observed value. The software generated value was comprised of 20% oil, 40% Smix, and 30 s sonication time. The predicted responses for droplet size, PDI, and Transmittance were 101 nm, 0.29, and 98.17% respectively. The observed response was 98.88 nm droplet size, 0.32 PDI, and 98.12% transmittance. The predicted and observed values were in good correlation, as seen in , establishing the accuracy of BDQ-F-SNEDDS.
2.5. Characterization
3. Materials
Bedaquiline fumarate (BDQ-F) (99.5% pure) was obtained as a complimentary sample from Omgene Life Sciences Pvt Ltd. (Vadodara, India). The high-performance liquid chromatography (HPLC) grade solvents, such as acetonitrile, methanol, and ethanol, were procured from Merck Ltd. in Mumbai, India. Transcutol-P and propylene glycol were acquired from Sigma-Aldrich (Bangalore, India). All other chemicals used in the study were of analytical grade, and deionized water was used for all experiments.
4. Methods
4.1. HPLC Method
The HPLC technique for analyzing BDQ was established following a documented procedure. In brief, an HPLC system (Shimadzu VP, Kyoto, Japan) was employed, featuring a C-18 column (250 × 4.6 mm, with 5 μm particle size), a binary pump, a UV-VIS detector, and Class VP software for data analysis. The mobile phase consisted of a mixture of water (0.01% TFA) and acetonitrile (0.01% TFA) in a ratio of 10:90 v/v, utilizing analytical grade solvents and filtered through a 0.45 μm membrane filter. The flow rate of the mobile phase was set at 1 mL/min, and the detector was configured to a wavelength of 265 nm [34]. This HPLC methodology served for assessing both the entrapment efficiency and in vitro drug release.
4.2. Solubility Studies
4.3. Pseudo-Ternary Phase Diagram
A pseudoternary phase diagram was built for the SNEDDS development to compute the surfactant: co-surfactant (Smix) and oil: Smix ratios. Smix was taken in the following ratios: 1:0, 1:1, 1:2, 2:1, 3:1, 4:1, 5:1. The oil and Smix mixtures were then titrated against deionized water. The amount of deionized water was changed at 5% intervals between 5 and 95%. After adding deionized water to the mixture, it was vortexed for 2 to 5 min and visually examined for clarity or turbidity, with the results noted on the phase diagram. The Smix, oil, and deionized water phases of the formulation are represented by the three vertices of the pseudoternary phase diagram. In the pseudoternary phase diagram, the percentage composition of each nanoemulsion was designated as a point, and the area surrounded by these points was measured [36].
4.4. Quality by Design (QbD) Approach Incorporation
Designing for quality gives value and quality to the final pharmaceutical product. Throughout the formulation development process, it is vital to identify and regulate the critical parameters related to the process and formulation development. The QbD technique entails establishing the quality target product profile (QTPP); identifying critical quality attributes (CQAs), critical material attributes (CMAs), and critical process parameters (CPPs) through screening and risk assessment; optimizing data using DoE; and evaluating the results. provide various parameters of QTPP and CQAs for SNEDDs formulation.
4.5. Risk Assessment Using Ishikawa
A risk assessment approach was developed to detect and estimate the likelihood of drug excipient interactions with various unit functions if there are any risks or failures. The Ishikawa fish-bone diagram was constructed employing Minitab 16 software, as shown in .
4.6. Formulation and Optimization of SNEDDS Using Box Behnken Design
Box–Behnken experimental design (BBD) (Design Expert® software version 10.0.4 by Stat-Ease in Minneapolis, Kansas, KS, USA) was used to evaluate the critical experimental conditions for the maximum and minimum response. BBD was used to optimize solid-nanoemulsion drug delivery systems (SNEDDS) by studying the effects of variables on size, entrapment efficiency, and drug release. This design helps to eliminate non-significant factors that do not have a significant effect on the response variable, and it can provide an optimized formulation with fewer trials, consequently leading to cost and time savings. Furthermore, this design simplifies the experiments by reducing the number of variables that need to be considered [37]. SNEDDSs were created after selecting the surfactant and co-surfactant ratio (Smix). Using a vortex, an exactly weighed quantity of drug was entirely dissolved in oil. Smix was slowly added drop by drop to the oil drug combination while constantly swirling to generate uniform formulation. After that, the mixture was sonicated to achieve the desired droplet size. BBD is a more suitable approach for evaluating the effects of formulation variables and the effect on their corresponding variables [38]. The goal of this optimization was to investigate how different independent variables influenced dependent variables. These factors included caprylic acid (A) used as an oil, ranging from 10 to 30 mL; Smix (B), consisting of propylene glycol as a surfactant, and Transcutol-P as a co-surfactant, ranging from 40 to 60 mL. The sonication time varied from 30 to 60 s. The dependent variables were the size of droplets (measured in nanometers), the polydispersity index (PDI), and the percentage of transmittance. The levels of independent variables, as well as the constraints of dependent variables, are presented in . Fourteen runs were generated with two center points with a quadratic polynomial equation.
4.7. Characterization of SNEDDS
4.8. In Vitro Drug Release Study
A drug release study was performed using a dialysis membrane. A pre-activated dialysis membrane with a molecular weight cut-off of 12.4 kDa was loaded with 2 mL of optimized BDQ-F-SNEDDS and BDQ-F suspension, respectively. Two separate 100 mL beakers were filled with 48 mL of 0.1 N HCl medium. The dialysis membrane containing the BDQ-F-SNEDDS and BDQ-F suspension was placed in the respective beakers filled with the medium. The beakers were then placed on a magnetic stirrer at a temperature of 37 °C and a stirring speed of 100 rpm.
Sampling was carried out at specific time intervals (2, 4, 6, 8, 10, and 12 h) by withdrawing 1 mL of sample at each time point. The withdrawn sample volume was replaced with an equal volume of fresh media to maintain sink conditions. HPLC analysis was performed on the collected samples to determine the drug concentration. The cumulative amount of drug release was calculated based on the analysis results. To ensure reliable data, the experiment was conducted in triplicate for statistical significance [41].
4.9. Stability Studies
The Stability studies were performed according to ICH guidelines [42]. The BDQ-F-loaded SNEDDS carriers were subjected to a physical stability study by storing them for the duration of 6 months under both room temperature and accelerated conditions. At predetermined intervals (0, 3, and 6 months), samples were taken from the storage and analyzed for any observable changes in Physical Appearance, droplet size, PDI, and entrapment efficiency. Additionally, the shelf-life of the optimized BDQ-F-SNEDDS was determined through this analysis [43]. Stability protocol is provided in as per industry format.
4.10. Cell Toxicity Studies
The cytotoxicity of both free BDQ-F and optimized BDQ-F-SNEDDS (self-nanoemulsifying drug delivery system) on A549 cells was assessed using the MTT assay. A549 cells were seeded at a density of (5 × 103) cells per well in 96-well tissue culture plates and incubated overnight. Subsequently, the cells were treated with varying concentrations (1–100 µg/mL) of the optimized BDQ-F-SNEDDS, placebo, or BDQ-F. Following the treatment, MTT was added to the wells, and the resulting formazan crystals were solubilized using dimethyl sulfoxide. The absorbance of the solution was measured [44]. The percentage of cytotoxicity, indicating cell viability, was calculated using the provided formula:
4.11. Statistical Analysis
The experiments were conducted in triplicate, and the data are presented as mean ± standard deviation (SD), with statistical significance set at p < 0.05. Data analysis was performed using GraphPad Prism® software, version 6.01.
5. Conclusions
Optimized BDQ-F-SNEDDSs were formulated using Design of Experiments (DoE), with Caprylic acid as the lipid and Transcutol P and propylene glycol as the surfactant/cosurfactant system, forming an interfacial film around the oil globules. The prepared SNEDDSs underwent evaluation for various parameters, including droplet size, PDI, and percent transmittance. Key Quality Target Product Profiles (QTPPs) and CQAs were identified to ensure a robust formulation. Risk assessment was performed using an Ishikawa Fish-bone diagram. Optimization of BDQ-F-SNEDDS was accomplished using the Box–Behnken design, resulting in an optimized formulation with a small droplet size and low PdI, indicating stability and robustness. Transmission electron microscopy (TEM) images exhibited a spherical shape and a distinct boundary, suggesting high entrapment efficiency.
Results from in vitro drug release studies have indicated that optimized BDQ-F-SNEDDS displayed greater concentrations of drug release compared to the suspension form. This suggests that the optimized BDQ-F-SNEDDS formulation effectively releases the drug in higher concentrations.
Cell cytotoxicity studies conducted on A549 cells revealed enhanced cellular internalization of BDQ-F-SNEDDS, which may be correlated with improved permeability. The lipophilic oil globules in SNEDDS could potentially enable its penetration through bacterial biofilms, thereby enhancing the effectiveness against resistant strains of bacteria. Furthermore, stability studies indicated that BDQ-F-SNEDDS remained stable at both 40 ± 2 °C and 25 ± 2 °C for up to 6 months.
The advantages of SNEDDS include its smaller globule size, which facilitates improved drug delivery. Furthermore, it also offers potential benefits for the long-term treatment of multidrug-resistant tuberculosis (MDR-TB) by extending the circulation time of the drug and providing a controlled release pattern. These characteristics hold promise for reducing the dose frequency of administration and enhancing patient compliance.
In summary, BDQ-F-SNEDDS presents a promising strategy for drug delivery that requires further exploration to optimize the treatment protocol for multidrug-resistant tuberculosis (MDR-TB) by decreasing the frequency of dosing and treatment duration. However, it is important to conduct additional research to fully understand the intricate in vivo disposition and pharmacodynamics in animal models.
References
- Global Tuberculosis Report 2020. 2020. Available online: https://www.who.int/publications/i/item/97892400131 (accessed on 25 September 2023).
- Ndjeka, N.; Schnippel, K.; Master, I.; Meintjes, G.; Maartens, G.; Romero, R.; Conradie, F. High treatment success rate for multidrug-resistant and extensively drug-resistant tuberculosis using a bedaquiline-containing treatment regimen. Eur. Respir. J. 2018, 52, 1801528. [Google Scholar] [CrossRef] [PubMed]
- Mencarini, J.; Spinicci, M.; Zammarchi, L.; Bartoloni, A. Tuberculosis in the European Region. Curr. Trop. Med. Rep. 2023, 10, 88–93. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 2 October 2023).
- Yadav, S. Grade III Severe QT Prolongation in an Indian Male on All-Oral Longer Regimen for Multidrug-Resistant Pulmonary Tuberculosis: World’s First Case. Cureus 2022, 14, e31819. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Dheda, K.; Chesov, D.; Mandalakas, A.M.; Udwadia, Z.; Horsburgh, C.R. Management of drug-resistant tuberculosis. Lancet 2019, 394, 953–966. [Google Scholar] [CrossRef]
- Rawal, T.; Butani, S. Combating tuberculosis infection: A forbidding challenge. Indian J. Pharm. Sci. 2016, 78, 8. [Google Scholar] [CrossRef]
- Ginsberg, A.M.; Spigelman, M. Challenges in tuberculosis drug research and development. Nat. Med. 2007, 13, 290–294. [Google Scholar] [CrossRef]
- Cox, E.; Laessig, K. FDA approval of bedaquiline—The benefit–risk balance for drug-resistant tuberculosis. N. Engl. J. Med. 2014, 371, 689–691. [Google Scholar] [CrossRef]
- Chahine, E.B.; Karaoui, L.R.; Mansour, H. Bedaquiline: A novel diarylquinoline for multidrug-resistant tuberculosis. Ann. Pharmacother. 2014, 48, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Courbon, G.M.; Bueler, S.A.; Mai, J.; Liu, J.; Rubinstein, J.L. Structure of mycobacterial ATP synthase with the TB drug bedaquiline. bioRxiv 2020. bioRxiv:2020.08.06.225375. [Google Scholar]
- Ibrahim, M.; Andries, K.; Lounis, N.; Chauffour, A.; Truffot-Pernot, C.; Jarlier, V.; Veziris, N. Synergistic Activity of R207910 Combined with Pyrazinamide against Murine Tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1011–1015. [Google Scholar] [CrossRef]
- Diacon, A.H.; Donald, P.R.; Pym, A.; Grobusch, M.; Patientia, R.F.; Mahanyele, R.; McNeeley, D.F. Randomized Pilot Trial of Eight Weeks of Bedaquiline (TMC207) Treatment for Multidrug-Resistant Tuberculosis: Long-Term Outcome, Tolerability, and Effect on Emergence of Drug Resistance. Antimicrob. Agents Chemother. 2012, 56, 3271–3276. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Martins, S.; Ferreira, D.; Segundo, M.A.; Reis, S. Lipid nanoparticles for topical and transdermal application for alopecia treatment: Development, physicochemical characterization, and in vitro release and penetration studies. Int. J. Nanomed. 2014, 9, 1231. [Google Scholar]
- Kakkar, A.K.; Dahiya, N. Bedaquiline for the treatment of resistant tuberculosis: Promises and pitfalls. Tuberculosis 2014, 94, 357–362. [Google Scholar] [CrossRef]
- Singh, B.; Bandopadhyay, S.; Kapil, R.; Singh, R.; Katare, O.P. Self-emulsifying drug delivery systems (SEDDS): Formulation development, characterization, and applications. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 427–521. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Singh, B.; Kumar, R.; Ahuja, N. Optimizing drug delivery systems using systematic design of experiments. Part I: Fundamental aspects. Crit. Rev. Ther. Drug Carr. Syst. 2005, 22. [Google Scholar] [CrossRef]
- Beg, S.; Sandhu, P.S.; Batra, R.S.; Khurana, R.K.; Singh, B. QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug Deliv. 2015, 22, 765–784. [Google Scholar] [CrossRef]
- Singh, B.; Singh, R.; Bandyopadhyay, S.; Kapil, R.; Garg, B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf. B Biointerfaces 2013, 101, 465–474. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.P.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef]
- Dhawan, S.; Kapil, R.; Singh, B. Formulation development and systematic optimization of solid lipid nanoparticles of quercetin for improved brain delivery. J. Pharm. Pharmacol. 2011, 63, 342–351. [Google Scholar] [CrossRef]
- Raza, K.; Singh, B.; Singal, P.; Wadhwa, S.; Katare, O.P. Systematically optimized biocompatible isotretinoin-loaded solid lipid nanoparticles (SLNs) for topical treatment of acne. Colloids Surf. B Biointerfaces 2013, 105, 67–74. [Google Scholar] [CrossRef]
- Singh, B.; Mehta, G.; Kumar, R.; Bhatia, A.; Ahuja, N.; Katare, O. Design, development and optimization of nimesulide-loaded liposomal systems for topical application. Curr. Drug Deliv. 2005, 2, 143–153. [Google Scholar] [CrossRef]
- Shahba, A.A.-W.; Mohsin, K.; Alanazi, F.K.; Abdel-Rahman, S.I. Optimization of self-nanoemulsifying formulations for weakly basic lipophilic drugs: Role of acidification and experimental design. Braz. J. Pharm. Sci. 2016, 52, 653–667. [Google Scholar] [CrossRef]
- Solans, C.; Izquierdo, P.; Nolla, J.; Azemar, N.; Garcia-Celma, M.J. Nano-emulsions. Curr. Opin. Colloid Interface Sci. 2005, 10, 102–110. [Google Scholar] [CrossRef]
- Limbani, M.D.; Patel, L. Studies on drug solubilization and role of lipid vehicle in pseudo ternary phase diagram in formulation development of SNEDDS containing poorly water soluble drug. Int. J. Pharm. Sci. Rev. Res. 2016, 40, 228–237. [Google Scholar]
- Marasini, N.; Yan, Y.D.; Poudel, B.K.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development and Optimization of Self-Nanoemulsifying Drug Delivery System with Enhanced Bioavailability by Box–Behnken Design and Desirability Function. J. Pharm. Sci. 2012, 101, 4584–4596. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Baskaran, R.; Balakrishnan, P.; Thapa, P.; Yong, C.S.; Yoo, B.K. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein. Eur. J. Pharm. Biopharm. 2011, 79, 250–257. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, R.; Espinosa-Andrews, H.; Morales-Hernández, N.; Lobato-Calleros, C.; Vernon-Carter, E.J. Mesquite gum/chitosan insoluble complexes: Relationship between the water state and viscoelastic properties. J. Dispers. Sci. Technol. 2019, 40, 1345–1352. [Google Scholar] [CrossRef]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Aldawsari, H.M.; Hosny, K.M.; Ahmad, J.; Akhter, S.; Kammoun, A.K.; Md, S. Formulation design and pharmacokinetic evaluation of docosahexaenoic acid containing self-nanoemulsifying drug delivery system for oral administration. Nanomater. Nanotechnol. 2020, 10, 1847980420950988. [Google Scholar] [CrossRef]
- Shukla, S.K.; Kulkarni, N.S.; Farrales, P.; Kanabar, D.D.; Parvathaneni, V.; Kunda, N.K.; Gupta, V. Sorafenib Loaded Inhalable Polymeric Nanocarriers against Non-Small Cell Lung Cancer. Pharm. Res. 2020, 37, 67. [Google Scholar] [CrossRef] [PubMed]
- Maloney, S.E.; Stewart, I.E.; Podell, B.K.; Gary, H.E.; Mecham, J.B.; Berube, B.J.; Hickey, A.J. Preparation Strategies of the Anti-Mycobacterial Drug Bedaquiline for Intrapulmonary Routes of Administration. Pharmaceuticals 2023, 16, 729. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, L.; Tilton, S.; Wang, J. Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J. Pharm. Sci. 2007, 96, 3052–3071. [Google Scholar] [CrossRef]
- Czajkowska-Kośnik, A.; Szekalska, M.; Amelian, A.; Szymańska, E.; Winnicka, K. Development and Evaluation of Liquid and Solid Self-Emulsifying Drug Delivery Systems for Atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. DoE based Olanzapine loaded poly-caprolactone nanoparticles decreases extrapyramidal effects in rodent model. Int. J. Pharm. 2018, 541, 198–205. [Google Scholar] [CrossRef]
- Yadav, P.; Rastogi, V.; Verma, A. Application of Box–Behnken design and desirability function in the development and optimization of self-nanoemulsifying drug delivery system for enhanced dissolution of ezetimibe. Future J. Pharm. Sci. 2020, 6, 7. [Google Scholar] [CrossRef]
- Usta, D.Y.; Timur, B.; Teksin, Z.S. Formulation development, optimization by Box-Behnken design, characterization, in vitro, ex-vivo, and in vivo evaluation of bosentan-loaded self-nanoemulsifying drug delivery system: A novel alternative dosage form for pulmonary arterial hypertension treatment. Eur. J. Pharm. Sci. 2022, 174, 106159. [Google Scholar] [PubMed]
- Syukri, Y.; Martien, R.; Lukitaningsih, E.; Nugroho, A.E. Novel Self-Nano Emulsifying Drug Delivery System (SNEDDS) of andrographolide isolated from Andrographis paniculata Nees: Characterization, in-vitro and in-vivo assessment. J. Drug Deliv. Sci. Technol. 2018, 47, 514–520. [Google Scholar] [CrossRef]
- Balakumar, K.; Raghavan, C.V.; Selvan, N.T.; Prasad, R.H.; Abdu, S. Self nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf. B Biointerfaces 2013, 112, 337–343. [Google Scholar] [CrossRef]
- Chinchole, A.; Poul, B.; Panchal, C.; Chavan, D. A review on stability guidelines by ICH and USFDA guidelines for new formulation and dosage form. PharmaTutor 2014, 2, 32–53. [Google Scholar]
- Singh, S.K.; Dadhania, P.; Vuddanda, P.R.; Jain, A.; Velaga, S.; Singh, S. Intranasal delivery of asenapine loaded nanostructured lipid carriers: Formulation, characterization, pharmacokinetic and behavioural assessment. RSC Adv. 2016, 6, 2032–2045. [Google Scholar] [CrossRef]
- Patil, S.M.; Sawant, S.S.; Kunda, N.K. Pulmonary delivery of bedaquiline-loaded cubosomes for non-small cell lung cancer (NSCLC) treatment. Drug Deliv. Lungs 2021, 32. [Google Scholar]